INTRODUCTION
SUMMARY
Autoimmune hemolytic anemia (AHA) is characterized by shortened red blood cell (RBC) survival and the presence of autoantibodies directed against autologous RBCs. Demonstration of antibody and/or complement on RBC membranes, usually by a positive direct antiglobulin test (DAT, also referred to as the Coombs test) is essential for diagnosis. Most patients with AHA (80 percent) exhibit warm-reactive antibodies of the immunoglobulin (Ig) G isotype on their red cells. Most of the remainder of patients exhibit cold-reactive autoantibodies. Two types of cold-reactive autoantibodies to RBCs are recognized: cold agglutinins and cold hemolysins. Cold agglutinins are generally of IgM isotype, whereas cold hemolysins usually are of IgG isotype. The DAT may detect IgG, proteolytic fragments of complement (mainly C3), or both on the RBCs of patients with warm-antibody AHA. In cold-antibody AHA, only complement is detected because the antibody dissociates from the RBCs during washing of the cells. About half of patients with AHA have no underlying associated disease; these cases are termed primary or idiopathic. Secondary cases are associated with underlying autoimmune, malignant, or infectious diseases or with ingestion of certain drugs.
Although most patients do not require transfusion of RBCs, transfusion should not be withheld from those with symptomatic anemia. In warm-antibody AHA, rituximab and glucocorticoids are effective in slowing the rate of hemolysis. Splenectomy is indicated for patients who are refractory to medical therapy or who require an unacceptably high maintenance dose or prolonged administration of glucocorticoids. Intravenous immunoglobulin may provide short-term control of hemolysis. Immunosuppressive drugs and danazol have been used successfully in refractory cases. In cold agglutinin- and cold hemolysin-mediated hemolysis, keeping the patient warm and treating underlying lymphoproliferative disorders usually are effective. Rituximab has been effective in about half of cases of cold AHA. Drug-induced immune hemolytic anemia usually is ameliorated by discontinuation of the offending drug.
Acronyms and Abbreviations:
AHA, autoimmune hemolytic anemia; CLL, chronic lymphocytic leukemia; DAF, decay-accelerating factor; DAT, direct antiglobulin test; HLA, human leukocyte antigen; HRF, homologous restriction factor; HS, hereditary spherocytosis; IAT, indirect antiglobulin test; Ig, immunoglobulin; IGHV, immunoglobulin heavy chain variable region; PNH, paroxysmal nocturnal hemoglobinuria; RBC, red blood cell; SLE, systemic lupus erythematosus.
DEFINITION AND HISTORY
The two main features of immune red blood cell (RBC) injury are (1) shortened RBC survival in vivo and (2) evidence of host antibodies reactive with autologous RBCs, most frequently demonstrated by a positive direct antiglobulin test (DAT), also known as the Coombs test. Most cases in adults are mediated by warm-reactive autoantibodies. A smaller proportion of patients exhibit cold-reactive autoantibodies or drug-related antibodies.
By the early 20th century, reticulocytes, spherocytes, and osmotic fragility of RBCs had been described. Clinicians could diagnose hemolytic anemia, but the distinction between congenital and acquired forms was imprecise. Some clinicians even doubted the existence of acquired hemolytic anemia.1 The sera of some patients with hemolytic anemia directly agglutinated saline suspensions of normal or autologous human RBCs. These serum factors, later shown to be specific antibodies (largely of the immunoglobulin [Ig] M class), were termed direct or saline agglutinins. In a smaller proportion of cases, the patients’ sera could mediate lysis of the test RBCs in the presence of fresh serum as a complement source. The heat-stable factors (antibodies) necessary for in vitro complement-mediated lysis were called hemolysins. However, in the majority of cases of hemolytic anemia, neither direct agglutinins nor hemolysins could be demonstrated. In 1945, Coombs and colleagues2 reported that RBCs coated with nonagglutinating Rh antibodies (now known to be of the IgG isotype) could be agglutinated by rabbit antiserum to human γ-globulin. That is, the rabbit antiglobulin serum crosslinked IgG antibody-coated RBCs to produce visible agglutination. Addition of rabbit antiglobulin serum to a suspension of washed RBCs isolated from patients with suspected acquired hemolytic anemia produced agglutination in many cases, including those patients lacking saline agglutinins or hemolysins. RBCs from patients with congenital hemolytic anemia did not agglutinate.3,4 This procedure now is termed the direct antiglobulin (Coombs) test. Subsequent studies established that positive direct antiglobulin reactions in autoimmune hemolytic anemia (AHA) are attributable to coating of the RBCs with immunoglobulins (mainly IgG) and/or complement proteins. When the RBCs are coated chiefly with complement proteins, a positive DAT depends upon the presence of anticomplement (principally anti-C3) in the antiglobulin reagent. In warm-antibody AHA, the autoantibodies, chiefly of IgG isotype, bind optimally to RBCs at 37°C. Warm antibodies may or may not activate complement binding to RBCs.
Cryopathic hemolytic syndromes are caused by autoantibodies that bind RBCs optimally at temperatures less than 37°C and usually less than 31°C. Two major types of “cold antibody” may produce AHA. Cold agglutinins, which directly agglutinate RBCs, mediate cold agglutinin disease. The Donath-Landsteiner autoantibody, which is not an agglutinin but a potent hemolysin, mediates paroxysmal cold hemoglobinuria. In both cryopathic syndromes, the complement system plays a major role in RBC injury (Chap. 19); as such, much greater potential exists for direct intravascular hemolysis than in warm-antibody–mediated AHA.
Cold agglutinins were first described by Landsteiner5 in 1903. However, recognition of the connection among cold agglutinins, hemolytic anemia, and Raynaud-like peripheral vascular phenomena evolved slowly. In 1918, Clough and Richter6 detected cold agglutinins in a patient with pneumonia. In 1925 and 1926, Iwai and MeiSai7,8 reported two patients with cold agglutinins and Raynaud phenomenon and showed that flow of blood through capillary tubes in vitro or in superficial capillaries in vivo was impeded at low temperatures. During the late 1940s and early 1950s, the observations of many investigators gradually established the pathogenic importance of cold agglutinins in RBC injury. Schubothe9 introduced the term cold agglutinin disease in 1953 and clearly distinguished the disorder from other acquired hemolytic syndromes.
In current usage, cold agglutinin disease pertains to patients with chronic AHA in which the autoantibody directly agglutinates human RBCs at temperatures below body temperature, maximally at 0 to 5°C. Fixation of complement to a patient’s RBCs by cold agglutinins in vivo occurs at higher temperatures but generally less than 37°C. Cold agglutinins typically are IgM, although occasionally they may be immunoglobulins of other isotypes. The cold agglutinins in chronic cold agglutinin disease generally are monoclonal. Most cold agglutinins have specificity for oligosaccharide antigens (I or i) of the RBC (see “Origin of Cold Agglutinins” below).
Donath and Landsteiner first described the cold hemolysin that bears their name in 1904. The Donath-Landsteiner antibody is responsible for complement-mediated hemolysis in paroxysmal cold hemoglobinuria, a rare form of AHA in adults. The disorder is characterized by recurrent episodes of massive hemolysis following cold exposure.10,11 A related form of hemolytic anemia occurs much more commonly in children (or young adults) as an acute, self-limited hemolytic process following several types of viral syndromes.10,11,12,13,14,15,16 The disease was recognized during the latter half of the 19th century, when the disease was more common because of its association with congenital or tertiary syphilis. With the advent of effective therapy for syphilis, this cause of paroxysmal cold hemoglobinuria has almost disappeared. Now, recurrent paroxysmal cold hemoglobinuria occurs very rarely in a chronic idiopathic form.10,11 An increasing proportion of Donath-Landsteiner autoantibody-mediated hemolytic anemias occurs as a single postviral episode in children, without recurrent attacks (paroxysms). The prognosis for such cases is excellent. Thus, rather than paroxysmal cold hemoglobinuria, a proposed term for this latter entity is Donath-Landsteiner hemolytic anemia.13,14
The first example of drug-related immune blood cell destruction was Ackroyd’s description of sedormid purpura in 1949.17 In 1953, Snapper and coworkers18 described a case of immune hemolysis and pancytopenia in a patient treated with mephenytoin (Mesantoin). Hemolysis ceased upon withdrawal of the drug. In 1956, Harris19 reported what are now classic studies of a patient who developed immune hemolytic anemia during a second course of stibophen administered for treatment of schistosomiasis. Since then, many drugs have been implicated in the production of positive DATs and accelerated RBC destruction.
AHA can be classified in two complementary ways (Table 54–1). The majority of cases (80 to 90 percent in adults) are mediated by warm-reactive autoantibodies10,11,20 or antibodies displaying optimal reactivity with human RBCs at 37°C. A smaller proportion of cases is attributable to cold-reactive autoantibodies exhibiting greater affinity for RBCs at temperatures less than 37°C. The distinction is important, not only because of differences in the pathophysiology of RBC injury but also in the therapeutic approaches required. An even smaller proportion of patients with AHA exhibit both cold-reactive and warm-reactive autoantibodies,21,22 which apparently recognize different antigens on the RBC membrane.23 RBC destruction is generally more severe in mixed cases.
|
Classification of AHA based on the presence or absence of underlying diseases also is useful (see Table 54–1). When no recognizable underlying disease is present, the AHA is termed primary or idiopathic. When AHA appears to be a manifestation or complication of an underlying disorder, the term secondary AHA is applied. Lymphocytic malignancies, particularly chronic lymphocytic leukemia (CLL) and lymphomas, account for approximately half of all secondary AHA cases and for the majority of AHA cases mediated by cold agglutinins.24 Systemic lupus erythematosus (SLE) and other autoimmune diseases account for a lesser but considerable proportion of secondary AHA cases. A large proportion of patients with mixed cold and warm autoantibodies have SLE.21,22 Infectious mononucleosis and Mycoplasma pneumoniae occasionally are associated with cryopathic AHA. Despite the frequent occurrence of immune thrombocytopenia and positive DATs in patients infected with HIV, AHA is relatively rare in these patients.25,26,27 Table 54–1 lists other associated diseases that are less-commonly reported. The etiologic and pathogenic significance of these associations is poorly understood, but most of the associated diseases involve components of the immune system, either by neoplasia or by aberrant immunopathologic responses.
Certain drugs also mediate immune injury to RBCs, and three general mechanisms are recognized (see Table 54–1 and Fig. 54–1). This classification is based on the effector mechanism of RBC injury, because the induction mechanism for formation of drug-related RBC antibodies is unknown. Two of the mechanisms, hapten-drug adsorption and ternary complex formation, involve drug-dependent antibodies. In the third mechanism, the drugs in question appear to induce formation of true autoantibodies capable of reacting with human RBCs in the absence of the inciting drug. These types of drug-mediated immune injury to RBCs often are referred to collectively as drug-immune hemolytic anemia to distinguish them from de novo AHA. Distinguishing among the mechanisms is not always possible, and some cases involve a combination of mechanisms. In addition, drug-related non-immunologic protein adsorption by RBCs may result in a positive DAT without actual RBC injury. This phenomenon should be distinguished from the three forms of drug-immune RBC injury. Table 54–2 lists drugs documented to cause either immune injury or a positive DAT.
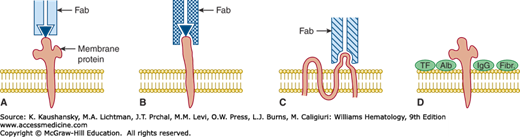
Figure 54–1.
Effector mechanisms by which drugs mediate a positive direct antiglobulin test. Relationships of drug, antibody-combining site, and red blood cell membrane protein are shown. Panels A, B, and C show only a single immunoglobulin Fab region (bearing one combining site). A. Drug adsorption/hapten mechanism. The drug (▾) binds avidly to an unknown red blood cell membrane protein in vivo. Antidrug antibody (usually immunoglobulin [Ig] G) binds to the protein-bound drug. The membrane protein is not known to be part of the epitope recognized by the antidrug antibody. The direct antiglobulin test (with anti-IgG) detects IgG antidrug antibody on the patient’s circulating (drug-coated) red blood cells. The indirect antiglobulin test detects antibody in the patient’s serum only when the test red blood cells have been previously coated with the drug by incubation in vitro. B. Ternary complex mechanism. Drug binds loosely or in undetectable amounts to red blood cell membrane. However, in the presence of appropriate antidrug antibody, a stable trimolecular (ternary) complex is formed by drug, red blood cell membrane protein, and antibody. In general, the antibody-combining site (Fab) recognizes both drug and membrane protein components but binds only weakly to either drug or protein unless both are present in the reaction mixture. In this mechanism, the direct antiglobulin test typically detects only red blood cell–bound complement components (e.g., C3 fragments) that are bound covalently and in large number to the patient’s red blood cells in vivo. The antibody itself escapes detection, possibly because of its low concentration but also because washing of the red cells (in the antiglobulin test procedure) apparently dissociates antibody and drug from the cells, leaving only the covalently bound C3 fragments. The indirect antiglobulin test also detects complement proteins on the test red blood cells when both antibody (patient serum) and a complement source (fresh patient serum or fresh normal serum) are present in the reaction mixture together with the drug. C. Autoantibody induction. Some drug-induced antibodies can bind avidly to red blood cell membrane proteins (usually Rh proteins) in the absence of the inducing drug and are indistinguishable from the autoantibodies of patients with autoimmune hemolytic anemia. The direct antiglobulin test detects the IgG antibody on the patient’s red blood cells. The indirect antiglobulin test usually detects antibody in the serum of patients with active hemolysis. D. Drug-induced nonimmunologic protein adsorption. Certain drugs cause plasma proteins to attach nonspecifically to the red blood cell membrane. The direct antiglobulin test detects nonspecifically bound IgG and complement components. If special antiglobulin reagents are used, other plasma proteins, such as transferrin (TF), albumin (Alb), and fibrinogen (Fibr), also may be detected. In contrast to the other mechanisms of drug-induced red blood cell injury, this mechanism does not shorten red blood cell survival in vivo.
| Drug | Reference | Drug | Reference |
|---|---|---|---|
| HAPTEN OR DRUG ADSORPTION MECHANISM | |||
| Penicillins | 28,29,30,31,32,33,34 | Tolbutamide | 44,45 |
| Cephalosporins | 35,36,37,38,39 | Cianidanol | 46 |
| Tetracycline | 40,41 | Hydrocortisone | 47 |
| 6-Mercaptopurine | 42 | Oxaliplatin | 48 |
| Carbromal | 43 | Cimetidine | 49 |
| TERNARY COMPLEX MECHANISM | |||
| Stibophen | 19 | Cephalosporins | 37,38,39,64 |
| Quinine | 50 | Diethylstilbestrol | 65 |
| Quinidine | 51,52 | Amphotericin B | 66 |
| Chlorpropamide | 53,54 | Doxepin | 67 |
| Metformin | 55 | Diclofenac | 68,69 |
| Rifampicin | 56 | Etodolac | 70 |
| Antazoline | 57 | Hydrocortisone | 47 |
| Thiopental | 58 | Oxaliplatin | 48 |
| Tolmetin | 59 | Pemetrexed | 71 |
| Probenecid | 60 | Cimetidine | 49 |
| Nomifensine | 61,62,63 | Iomeprol | 72 |
| AUTOANTIBODY MECHANISM | |||
| Cephalosporins | 39 | Latamoxef | 84 |
| Tolmetin | 59 | Glafenine | 84 |
| Nomifensine | 61 | Procainamide | 85 |
| α-Methyldopa | 73,74,75,76 | Diclofenac | 69,86 |
| L-dopa | 78,79,80,81 | Pentostatin | 87 |
| Mefenamic acid | 82,83 | Fludarabine | 88,89 |
| Teniposide | 84 | Cladribine | 90 |
| Oxaliplatin | 48 | Efalizumab | 91 |
| Cianidanol | 46 | Lenalidomide | 92 |
| NONIMMUNOLOGIC PROTEIN ADSORPTION | |||
| Cephalosporins | 93,94 | ||
| Oxaliplatin | 48 | ||
| Carboplatin | 48 | ||
| Cisplatin | 48,95 | ||
| UNCERTAIN MECHANISM OF IMMUNE INJURY | |||
| Mesantoin | 18 | Erythromycin | 106 |
| Phenacetin | 50 | 5-fluorouracil | 107 |
| Insecticides | 96 | Nalidixic Acid | 108 |
| Chlorpromazine | 97 | Sulindac | 109 |
| Melphalan | 98 | Omeprazole | 110 |
| Isoniazid | 99 | Temafloxacin | 111 |
| p-Aminosalicylic acid | 100 | Carboplatin | 112 |
| Acetaminophen | 101 | Efavirenz | 113 |
| Thiazides | 102 | Etoricoxib | 114 |
| Streptomycin | 103 | Levofloxacin | 115 |
| Ibuprofen | 104 | Pueraria (Chinese herb) | 116 |
| Triamterene | 105 | Cabergoline | 117 |
EPIDEMIOLOGY
The annual incidence of warm-antibody AHA is 1 per 75,000 to 80,000 population.11 Estimates of the frequency of primary (idiopathic) AHA vary from 20 to 80 percent of all types of AHA, depending on the referral patterns of the reporting center.11,20,118 In general, AHA is considered secondary (1) when AHA and the underlying disease occur together with greater frequency than can be accounted for by chance alone; (2) when the AHA reverses simultaneously with correction of the associated disease; or (3) when AHA and the associated disease are related by evidence of immunologic aberration.11 Using these criteria, the frequency of primary warm-antibody AHA probably is closer to 50 percent of all cases. Careful followup of patients with primary AHA is essential, because hemolytic anemia may be the presenting finding in a patient who subsequently develops overt evidence of an underlying disorder. For example, in one series, 18 of 107 patients with AHA developed a malignant lymphoproliferative disorder at a median of 26.5 months after diagnosis of the AHA.119
Warm-antibody AHA has been diagnosed in people of all ages, from infants to the elderly. The majority of patients are older than 40 years of age, with peak incidence around the seventh decade. This age distribution probably reflects, in part, the increased frequency of lymphoproliferative malignancies in the elderly, resulting in an age-related increase in the frequency of secondary AHA. Although multiple cases are occasionally observed in families,120,121,122 most cases of primary AHA arise sporadically. Development of AHA does not have an apparent association with any particular human leukocyte antigen (HLA) haplotype or other genetic factor.
Cold agglutinin disease is less common than warm-antibody AHA, with a prevalence of approximately 14 per 1 million population,24 accounting for only 10 to 20 percent of all cases of AHA.10,11,123 Women are affected more commonly than men.10,11 No genetic or racial factors are known to contribute to the pathogenesis of this disease.
Secondary cold agglutinin disease is seen most commonly in adolescents or young adults as a self-limited process associated with M. pneumoniae infections or infectious mononucleosis and, rarely, in children with chickenpox. The term also has been used to describe a chronic disorder occurring in older patients with known malignant lymphoproliferative diseases. On the other hand, idiopathic (primary) chronic cold agglutinin disease has its peak incidence after age 50 years. This disorder, with its characteristic monoclonal IgM cold agglutinins, may be considered a special form of monoclonal gammopathy (Chap. 106). Nearly all of these patients exhibit clonal B lymphocyte proliferation.24 As with other “essential” or idiopathic monoclonal gammopathies, some patients in this group gradually develop features of a B-cell lymphoproliferative disorder resembling Waldenström macroglobulinemia, CLL, or a B-cell lymphoma. Thus, the distinction between primary and secondary types of chronic cold agglutinin disease is not absolute.
Although the majority of patients with mycoplasma pneumonia have significant cold agglutinin titers, they only infrequently develop clinical hemolytic anemia.124,125,126 However, subclinical RBC injury may occur. In M. pneumoniae infections, weakly positive direct antiglobulin reactions and/or mild reticulocytosis are often noted in the absence of anemia in a substantial number of cases.124 Cold agglutinins occur in more than 60 percent of patients with infectious mononucleosis, but again, hemolytic anemia is rare.127,128,129
Medical centers that receive many referrals report that paroxysmal cold hemoglobinuria constitutes 2 to 5 percent of all cases of AHA.10,11 Among children, however, Donath-Landsteiner hemolytic anemia accounted for 32.4 percent of 68 immune hemolytic syndromes diagnosed over a 4-year period.15 Commonly, the diagnosis is missed because of lack of physician awareness or failure to perform the proper serologic studies (see “Serologic Features” below).12,15 Thus, the true incidence may be higher. Although familial occurrence has been reported, no racial or genetic risk factors are known.10 As noted, most childhood cases follow either specific viral infections or upper respiratory infections of undefined etiology.10,11,12,13,14,15
Older series report that drug-induced immune hemolytic anemia accounts for 12 to 18 percent of immune hemolytic anemias.11 The disorder is much less common now that α-methyldopa and megaunit doses of penicillin rarely are used. The current incidence of drug-induced immune hemolytic anemia is estimated at 1 per 1 million population, approximately 88 percent of which result from the second- and third-generation cephalosporins, cefotetan, and ceftriaxone.89,130 Fludarabine has replaced α-methyldopa as the most common cause of drug-induced autoantibodies.89
ETIOLOGY AND PATHOGENESIS
The etiology of AHA is unknown. In warm-antibody AHA, the autoantibodies that mediate RBC destruction are predominantly (but not exclusively) IgG globulins possessing relatively high binding affinity for human RBCs at 37°C. As a result, the major share of plasma autoantibody is bound to the patient’s circulating RBCs. Eluates prepared from the patient’s washed, autoantibody-coated RBCs constitute an important source of purified autoantibody for investigation of specificity, immunoglobulin structure, or other properties. In addition, sera from patients with warm AHA often are used in blood banks for crossmatching and for general screening of antibody specificity. The quantity of such autoantibody in serum may be low and in some cases may not reflect the full spectrum of anti-RBC specificity revealed in concurrently prepared RBC eluates.124
In patients with primary AHA, erythrocyte autoantibodies are the only recognizable immunologic aberration. Furthermore, the autoantibodies of any one patient often are specific for only a single RBC membrane protein (see “Serologic Features” below). The narrow spectrum of autoreactivity suggests the mechanism underlying AHA development in such patients is not secondary to a generalized defect in immune regulation. Rather, these patients may develop warm-antibody AHA through an aberrant immune response to a self-antigen or to an immunogen that mimics a self-antigen.
In patients with secondary AHA, the disease may be associated with a fundamental disturbance in the immune system, for example, when in the setting of lymphoma, CLL, SLE, primary agammaglobulinemia (common variable immunodeficiency), or hyper-IgM immunodeficiency syndrome. In these settings, warm-antibody AHA most likely arises through an underlying defect in immune regulation, although the contribution of an aberrant immune response to self-antigen cannot be excluded. AHA seems especially frequent in patients with low-grade lymphoma or CLL treated with fludarabine 88,89 or 2-chlorodeoxyadenosine (cladribine).90 The T-lymphocytopenia induced by these drugs may exacerbate the preexisting tendency of patients to form autoantibodies.
A long-recognized but poorly understood phenomenon, the development of AHA or a positive DAT following RBC transfusion, has received renewed interest lately.132,133 Although generally transient, the positive DAT may persist for up to 300 days in some transfusion recipients, long after any transfused RBCs have disappeared.134,135 It is not clear whether this represents true autoimmunity or some other mechanism, for example, microchimerism resulting from temporary engraftment of passenger memory lymphocytes from the RBC donor.132
A still unexplained observation is that certain drugs, such as α-methyldopa, can induce warm-reacting IgG anti-RBC autoantibodies in otherwise normal persons. The autoantibodies induced by α-methyldopa have Rh-related serologic and immunochemical136 specificity similar to that of autoantibodies arising in many patients with “spontaneous” AHA. A critical difference is that the drug-associated autoantibodies subside when the drug is discontinued, suggesting that (1) the latent potential to form this type of anti-RBC autoantibody is present in many immunologically normal individuals, and (2) the steps required to generate such autoantibodies do not necessarily create a sustained autoimmune state. On the other hand, maintenance of chronic idiopathic AHA may be either secondary to a continuing (but unknown) stimulus or induced by a short stimulus to which the patient continues to respond.
Normal subjects sometimes have a positive DAT when they volunteer to donate blood.137,138 The positive DAT in these normal donors often results from warm-reacting IgG autoantibodies, similar in serologic specificity131 and in IgG subclass137 to the autoantibodies occurring in AHA. Although many of these donors remain DAT-positive without developing overt hemolytic anemia, a few have been documented to develop AHA.137,138 The prevalence of positive DATs in normal blood donors is approximately 1 in 10,000 donors.137,139 Because blood donation per se likely does not contribute to an increased risk of developing autoantibodies, the 1 in 10,000 proportion likely is the approximate frequency of positive DATs in the entire population. A proportion of patients who present with clinically overt primary AHA may come from a subset of asymptomatic individuals who are DAT-positive, but this notion is not established.
Several concepts have been developed to explain immunologic tolerance to self-antigens.140,141,142,143 Relevant to warm-antibody AHA, membrane-bound antigens expressed in a multivalent array at high concentration may induce tolerance by effecting clonal deletion of autoreactive B cells.144 Both the Rh-related and the non-Rh types of RBC antigens targeted by AHA autoantibodies (see “Serologic Features” below) are expressed normally by human fetal erythrocytes, as early as 10 to 12 weeks of life.145 However, because new B cells develop daily in the marrow throughout life and because B cells may somatically mutate their Ig receptors, self-tolerance in the B cell compartment is never assured. Analogy to observations in NZB (New Zealand black) mice146,147 suggests the peritoneal cavity is a privileged compartment that shelters autoreactive B cells from host RBCs, allowing them to escape deletion, later to produce anti-RBC autoantibodies with appropriate T-cell help. The strong predominance of IgG antibodies in AHA suggests B-cell isotype switching, which is consistent with the idea of an antigen-driven process. Moreover, because T-cell help is necessary for inducing B-cell isotype switching, the pathway(s) to autoantibody induction in AHA also may involve an abnormal or unique mode of antigen presentation to T cells.148
A high proportion of monoclonal IgM cold agglutinins with either anti-I or anti-i specificity have heavy-chain variable regions encoded by IGHV4–34 (immunoglobulin heavy chain variable region), formerly designated IGHV4.21.143,149,150,151 IGHV4–34 encodes a distinct idiotype identified by the rat monoclonal antibody 9G4. This idiotype is expressed both by the cold agglutinins themselves and on the surface immunoglobulin of B cells synthesizing cold agglutinins or related immunoglobulins possessing IGHV4–34 sequences.152 Using the 9G4 monoclonal antibody as a probe, this idiotype was found not only in a very high proportion of circulating B cells and marrow lymphoplasmacytoid cells of patients with lymphoma-associated chronic cold agglutinin disease, but also in a smaller proportion of B cells in the blood and lymphoid tissues of normal adult donors and in the spleens of 15-week human fetuses.152 These data suggest B cells expressing the IGHV4–34 gene (or a closely related sequence) are present throughout ontogeny. Therefore, chronic cold agglutinin disease may represent a marked, unregulated expansion of a subset (clone) of such B cells.
Light-chain V-region gene use in anti-I cold agglutinins is highly selective. A strong bias toward use of the κ III variable region subgroup (Vκ-III) is observed.150,151,152,153 Light-chain selection among anti-i cold agglutinins, however, is much more variable and includes the λ type.150,151,152,153,154
Observations that pathologic cold agglutinins are synthesized with distinct and highly selected V-region sequences must be viewed against the background of two other subsequent observations. First, IGHV4–34 or related IGHV genes also may encode the heavy-chain variable regions of other types of antibodies, such as rheumatoid factor autoantibodies and alloantibodies to a variety of blood group antigens, including polypeptide determinants such as Rh.155 Second, normal human antibodies to an exogenous carbohydrate antigen, Haemophilus influenzae type b capsular polysaccharide, also are encoded by a restricted set of IGHV genes156 and Ig light-chain V genes.157 Thus, regulation of Ig gene use for production of anti-I or anti-i cold agglutinins may not differ fundamentally from normal antibody formation to other carbohydrate antigens.
In the setting of B-cell lymphoma or Waldenström macroglobulinemia, cold agglutinins may be produced by the malignant clone itself. Two patients with lymphoma and monoclonal cold agglutinin were identified as having a karyotypically abnormal B-cell clone that produced a cold agglutinin identical to that found in their sera.158,159 Trisomy 3 has been the most frequently observed karyotypic abnormality in patients with non-Hodgkin lymphoma and cold agglutinins.158,160
Normal human sera generally have naturally occurring polyclonal cold agglutinins in low titer (usually 1/64 or less).10 Otherwise healthy persons may develop elevated titers of cold agglutinins specific for I/i antigens during certain infections (e.g., M. pneumoniae, Epstein-Barr virus, cytomegalovirus). In contrast to other forms of cold agglutinin disease, hyperproduction of these postinfectious cold agglutinins is transient. Some evidence indicates postinfectious cold agglutinins may be less clonally restricted than those occurring in chronic cold agglutinin disease,161 but this finding is not universal.162 Whether IGHV4–34 also encodes most heavy-chain variable regions of all naturally occurring or postinfectious cold agglutinins remains to be determined.
The increased production of cold agglutinins in response to infection with M. pneumoniae may be secondary to the fact that the oligosaccharide antigens of the I/i type serve as specific Mycoplasma receptors.163 This process may lead to altered antigen presentation involving a complex between a self-antigen (I/i) and a non–self-antigen (Mycoplasma). Alternatively, the anti-i cold agglutinins may arise as a consequence of polyclonal B cell activation, as occurs in infectious mononucleosis (Chap. 82).
The mechanism(s) whereby dissimilar infectious agents (e.g., spirochetes and several types of virus) induce the immune system to produce Donath-Landsteiner antibodies with specificity for the human P blood group antigen (see “Serologic Features” below) is not known.
Warm autoantibodies to RBCs in AHA are pathogenic. In contrast to autologous RBCs, labeled RBCs lacking the antigen targeted by the autoantibodies may survive normally in patients with warm-antibody AHA.10,164,165 Furthermore, transplacental passage of IgG anti-RBC autoantibodies from a mother with AHA to the fetus can induce intrauterine or neonatal hemolytic anemia.166 Finally, despite notable exceptions and differences related to IgG subclass of the autoantibody, in general, an inverse relationship between the quantity of RBC-bound IgG antibody and RBC survival is noted in serial studies performed on animals and patients.167,168,169,170,171,172
In warm-antibody AHA, the patient’s RBCs typically are coated with IgG autoantibodies with or without complement proteins. Autoantibody-coated RBCs are trapped by macrophages in the Billroth cords of the spleen and, to a lesser extent, by Kupffer cells in the liver (Chap. 68).164,167,168,170,171,172,173,174 The process leads to generation of spherocytes and fragmentation and ingestion of antibody-coated RBCs.175,176 The macrophage has surface receptors for the Fc region of IgG, with preference for the IgG1 and IgG3 subclasses177,178 and surface receptors for opsonic fragments of C3 (C3b and C3bi) and C4b.179,180,181 When present together on the RBC surface, IgG and C3b/C3bi appear to act cooperatively as opsonins to enhance trapping and phagocytosis.170,171,180,181,182,183,184 Although RBC sequestration in warm-antibody AHA occurs primarily in the spleen,164,171,172,173 very large quantities of RBC-bound IgG167,168,174 or the concurrent presence of C3b on the RBCs167,170,171 may favor trapping in the liver.
Interaction of a trapped RBC with splenic macrophages may result in phagocytosis of the entire cell. More commonly, a type of partial phagocytosis results in spherocyte formation. As RBCs adhere to macrophages via the Fc receptors, portions of RBC membrane are internalized by the macrophage. Because membrane is lost in excess of contents, the noningested portion of the RBC assumes a spherical shape, the shape with the lowest ratio of surface area to volume.175,176,185 Spherical RBCs are more rigid and less deformable than normal RBCs. As such, spherical RBCs are fragmented further and eventually destroyed in future passages through the spleen. Spherocytosis is a consistent and diagnostically important hallmark of AHA,186 and the degree of spherocytosis correlates well with the severity of hemolysis.10
Direct complement-mediated hemolysis with hemoglobinuria is unusual in warm-antibody AHA, even though many warm autoantibodies fix complement. The failure of C3b-coated RBCs to be hemolyzed by the terminal complement cascade (C5–C9) has been attributed, at least in part, to the ability of complement regulatory proteins (factors I and H) in plasma and C3b receptors on the RBC surface to alter the hemolytic function of cell-bound C3b and C4b.187 Glycosylphosphatidylinositol-linked erythrocyte membrane proteins, such as decay-accelerating factor (DAF; CD55) and homologous restriction factor (HRF; CD59), may limit the action of autologous complement on autoantibody-coated RBCs.188,189,190 DAF inhibits the formation and function of cell-bound C3-converting enzyme,188 thus, indirectly limiting formation of C5-converting enzyme. HRF, on the other hand, impedes C9 binding and formation of the C5b–9 membrane attack complex.189
Cytotoxic activities of macrophages and lymphocytes also may play a role in the destruction of RBCs in warm-antibody AHA. Monocytes can lyse IgG-coated RBCs in vitro independently of phagocytosis.191,192 Cell-bound complement is neither necessary nor sufficient for such cytotoxicity, but bound C3b/C3d can potentiate the effects of IgG.192 In one study, cytotoxicity, but not phagocytosis, was inhibited by hydrocortisone in vitro.191 Lymphocytes also can lyse IgG antibody-coated RBCs in vitro.193,194,195 The relative contribution of antibody-dependent monocyte- and lymphocyte-mediated cytotoxicity to RBC destruction in patients with warm-antibody AHA is not known.
Most cold agglutinins are unable to agglutinate RBCs at temperatures higher than 30°C. The highest temperature at which these antibodies cause detectable agglutination is termed the thermal amplitude. The value varies considerably among patients. Generally, patients with cold agglutinins with higher thermal amplitudes have a greater risk for cold agglutinin disease.9 For example, active hemolytic anemia has been observed in patients with cold agglutinins of modest titer (e.g., 1:256) and high thermal amplitudes.196
The pathogenicity of a cold agglutinin depends upon its ability to bind host RBCs and to activate complement.10,182,197,198 This process is called complement fixation. Although in vitro agglutination of the RBCs may be maximal at 0 to 5°C, complement fixation by these antibodies may occur optimally at 20 to 25°C and may be significant at even higher physiologic temperatures.10,196,197 Agglutination is not required for the process. The great preponderance of cold agglutinin molecules are IgM pentamers, but small numbers of IgM hexamers with cold agglutinin activity are found in patients with cold agglutinin disease. Hexamers fix complement and lyse RBCs more efficiently than do pentamers, suggesting that hexameric IgM plays a role in the pathogenesis of hemolysis in these patients.199
Cold agglutinins may bind to RBCs in superficial vessels of the extremities, where the temperature generally ranges between 28 and 31°C, depending upon ambient temperature.200 Cold agglutinins of high thermal amplitude may cause RBCs to aggregate at this temperature, thereby impeding RBC flow and producing acrocyanosis. In addition, the RBC-bound cold agglutinin may activate complement via the classic pathway. Once activated complement proteins are deposited onto the RBC surface, the cold agglutinin need not remain bound to the RBCs for hemolysis to occur. Instead, the cold agglutinin may dissociate from the RBCs at the higher temperatures in the body core and again be capable of binding other RBCs at the lower temperatures in the superficial vessels. As a result, patients with cold agglutinins of high thermal amplitude tend toward a sustained hemolytic process and acrocyanosis.201 In contrast, patients with antibodies of lower thermal amplitude require significant chilling to initiate complement-mediated injury of RBCs. This sequence may result in a burst of hemolysis with hemoglobinuria.201 Combinations of these clinical patterns also occur. Cold agglutinins of the IgA isotype, an isotype that does not fix complement, may cause acrocyanosis but not hemolysis.202 Thus, the relative degree of hemolysis or impeded RBC flow is influenced significantly by the properties and quantity of the cold agglutinins in a given patient.
Complement fixation by cold agglutinins may affect RBC injury by two major mechanisms: (1) direct lysis and (2) opsonization for hepatic and splenic macrophages. Both mechanisms probably operate to varying degrees in any patient. Direct lysis requires propagation of the full C1 to C9 sequence on the RBC membrane. If this process occurs to a significant degree, the patient may experience intravascular hemolysis leading to hemoglobinemia and hemoglobinuria. Intravascular hemolysis of this severity is relatively rare because phosphatidylinositol-linked RBC membrane proteins (DAF and HRF) protect against injury by autologous complement components. Thus, the complement sequence on many RBCs is completed only through the early steps, leaving opsonic fragments of C3 (C3b/C3bi) and C4 (C4b) on the cell surface. The fragments provide only a weak stimulus for phagocytosis by monocytes in vitro.184,203 However, activated macrophages may ingest C3b-coated particles avidly.204 Accordingly, RBCs heavily coated with C3b (and/or C3bi) may be removed from the circulation by macrophages either in the liver or, to a lesser extent, the spleen.171,197,205,206 The trapped RBCs may be ingested entirely or released back into the circulation as spherocytes after losing plasma membrane.
In vivo studies of the fate of 51Cr-labeled C3b-coated RBCs170,197,205,206 indicate many of the erythrocytes trapped in the liver or spleen gradually may reenter the circulation. The released cells generally are coated with the opsonically inactive C3 fragment C3dg. Conversion of cell-bound C3b or C3bi to C3dg results from the action of the naturally occurring complement inhibitor factor I in concert with factor H or CR1 receptors.181 The surviving C3dg-coated RBCs circulate with a near-normal life span170,197,205,206 and are resistant to further uptake of cold agglutinins or complement.197,205,207 However, C3dg-coated RBCs also may react in vitro with anticomplement (anti-C3) serum in the DAT. In fact, most of the antiglobulin-positive RBCs of patients with cold agglutinin disease are coated with C3dg.
In paroxysmal cold hemoglobinuria, the mechanism of hemolysis probably parallels in vitro events (see “Serologic Features” below). During severe chilling, blood flowing through skin capillaries is exposed to low temperatures. The Donath-Landsteiner antibody and early acting complement components presumably bind to RBCs at the lowered temperatures. Upon return of the cells to 37°C in the central circulation, the cells are lysed by propagation of the terminal complement sequence through C9. The Donath-Landsteiner antibody itself dissociates from the RBCs at 37°C. Erythrocyte membrane proteins that restrict C5b–9 assembly (e.g., HRFs) may be less effective in controlling Donath-Landsteiner antibody-initiated complement activation than that initiated by cold agglutinins.
Table 54–3 summarizes the three mechanisms of drug-mediated immune injury to RBCs. Drugs also may mediate protein adsorption to RBCs by nonimmune mechanisms, but RBC injury does not occur.
| Hapten/Drug Adsorption | Ternary Complex Formation | Autoantibody Binding | Nonimmunologic Protein Adsorption | |
|---|---|---|---|---|
| Prototype drug | Penicillin | Quinidine | α-Methyldopa | Cephalothin |
| Role of drug | Binds to red cell membrane | Forms ternary complex with antibody and red cell membrane component | Induces formation of antibody to native red cell antigen | Possibly alters red cell membrane |
| Drug affinity to cell | Strong | Weak | None demonstrated to intact red cell but binding to membranes reported | Strong |
| Antibody to drug | Present | Present | Absent | Absent |
| Antibody class predominating | Immunoglobulin (Ig) G | IgM or IgG | IgG | None |
| Proteins detected by direct antiglobulin test | IgG, rarely complement | Complement | IgG, rarely complement | Multiple plasma proteins |
| Dose of drug associated with positive antiglobulin test | High | Low | High | High |
| Presence of drug required for indirect antiglobulin test | Yes (coating test red cells) | Yes (added to test medium) | No | Yes (added to test medium) |
| Mechanism of red cell destruction | Splenic sequestration of IgG-coated red cells | Direct lysis by complement plus splenic–hepatic clearance of C3b-coated red cells | Splenic sequestration | None |
This mechanism applies to drugs that can bind firmly to proteins, including RBC membrane proteins. The classic setting is very-high-dose penicillin therapy,28,29,30,31,32,33,34 which is encountered less commonly today than in previous decades.
Most individuals who receive penicillin develop IgM antibodies directed against the benzylpenicilloyl determinant of penicillin, but this antibody plays no role in penicillin-related immune injury to RBCs. The antibody responsible for hemolytic anemia is of the IgG class, occurs less frequently than the IgM antibody, and may be directed against the benzylpenicilloyl,31 or, more commonly, nonbenzylpenicilloyl determinants.28,29,30,32 Other manifestations of penicillin sensitivity usually are not present.
All patients receiving high doses of penicillin develop substantial coating of RBCs with penicillin. The penicillin coating itself is not injurious. If the penicillin dose is very high (10 to 30 × 106 units per day, or less in the setting of renal failure) and promotes cell coating, and if the patient has an IgG antipenicillin antibody, the antibody binds to the RBC-bound penicillin molecules and the DAT with anti-IgG becomes positive (see Fig. 54–1A).29,31,32,51,208 Antibodies eluted from patients’ RBCs or present in their sera react in the indirect antiglobulin test (IAT) only against penicillin-coated RBCs. This step is critical in distinguishing these drug-dependent antibodies from true autoantibodies.
Not all patients receiving high-dose penicillin develop a positive DAT reaction or hemolytic anemia because only a small proportion of such individuals produce the requisite antibody. Destruction of RBCs coated with penicillin and IgG antipenicillin antibody occurs mainly through sequestration by splenic macrophages.30,209 In some patients with penicillin-induced immune hemolytic anemia, blood monocytes and presumably splenic macrophages may lyse the IgG-coated RBCs without phagocytosis.210 Hemolytic anemia resulting from penicillin typically occurs only after the patient has received the drug for 7 to 10 days and ceases a few days to 2 weeks after the patient discontinues taking the drug.
Low-molecular-weight substances, such as drugs, generally are not immunogenic in their own right. Induction of antidrug antibody is thought to require firm chemical coupling of the drug (as a hapten) to a protein carrier. In the case of penicillin, the carrier protein involved in antibody induction need not be the same as the erythrocyte membrane protein to which penicillin is coupled in the effector phase, that is, when the IgG antipenicillin antibodies bind to penicillin-coated RBCs. In contrast to evidence on the ternary complex mechanism, no evidence indicates the drug-dependent antibodies responsible for RBC injury in this hapten/drug adsorption mechanism also recognize native erythrocyte membrane structures.
Cephalosporins have antigenic cross-reactivity with penicillin211,213 and bind firmly to RBC membranes, as do semisynthetic penicillins.33,34 Hemolytic anemia similar to that seen with penicillin has been ascribed to cephalosporins35,36,37,38,39 and some semisynthetic penicillins.33,34 Tetracycline40,41 and tolbutamide44,45 also may cause hemolysis by this mechanism. Carbromal causes positive IgG antiglobulin reactions by a similar mechanism,43 but hemolytic anemia has not been described.
Many drugs can induce immune injury not only of RBCs but also of platelets or granulocytes by a process that differs in several ways from the mechanism of hapten/drug adsorption (see Table 54–3). First, drugs in this group (see Table 54–2) exhibit only weak direct binding to blood cell membranes. Second, a relatively small dose of drug is capable of triggering destruction of blood cells. Third, cellular injury appears to be mediated chiefly by complement activation at the cell surface. The cytopathic process induced by such drugs previously has been termed the innocent bystander or immune complex mechanism. The terminology reflected the prevailing notion that, in vivo, drug–antibody complexes formed first (immune complexes) and then became secondarily bound to target blood cells as “innocent bystanders,” either nonspecifically or possibly via membrane receptors (e.g., Fcγ receptors on platelets or C3b receptors on red cells), with the potential for subsequent activation of complement by bound complexes.
The “immune complex” and “innocent bystander” terminology now seems less appropriate because of models developed from research on analogous drug-dependent platelet injury214,215,216 (Chap. 117) and a series of relevant serologic observations on drug-mediated immune hemolytic anemia. These studies suggest blood cell injury is mediated by a cooperative interaction among three reactants to generate a ternary complex (see Fig. 54–1B) involving (1) the drug (or drug metabolite in some cases), (2) a drug-binding membrane site on the target cell, and (3) antibody. For example, several patients possessed drug-dependent antibodies that exhibited specificity for RBCs bearing defined alloantigens such as those of the Rh, Kell, or Kidd blood groups. That is, even in the presence of drug, the antibodies were selectively nonreactive with human RBCs lacking the alloantigen in question.58,84,217,218,219 In each case, high-affinity drug binding to cell membrane could not be demonstrated. The drug-dependent antibody is thought to bind, through its Fab domain, to a compound neoantigen consisting of loosely bound drug and a blood group antigen intrinsic to the red cell membrane. Elegant studies on quinidine- or quinine-induced immune thrombocytopenia have demonstrated the IgG antibodies implicated in this disorder bind through their Fab domains, not by their Fc domains to platelet Fcγ receptors.220,221
The data elucidate how one patient with quinidine sensitivity may have selective destruction of platelets and another may have selective destruction of RBCs. This process occurs because the pathogenic antibody recognizes the drug only in combination with a particular membrane structure of the RBC (e.g., a known alloantigen) or of the platelet (e.g., α domain of the glycoprotein Ib complex). Therefore, at least in these cases, the target cell does not appear to be purely an innocent bystander. Binding of the drug itself to the target cell membrane is weak until the attachment of the antibody to both drug and cell membrane is stabilized. Yet the binding of the antibody is drug dependent. Such a three-reactant interdependent “troika” is unique to this mechanism of immune cytopenia.
The foregoing discussion depicting drugs as creating a “self + non-self” neoantigen on the target cell applies to the effector phase as opposed to the induction phase of the process. However, the same drug-binding membrane protein appears to be involved in forming the immunogen that induces the antibody, as evidenced by drug-dependent antibodies exhibiting selective reactivity with defined red cell alloantigens (carrier specificity).58,84,217,218,219 How this process is accomplished in the absence of evidence for strong, covalent binding of the drugs in this group to a host membrane protein remains to be elucidated.
RBC destruction by this mechanism may occur intravascularly after completion of the whole complement sequence, resulting in hemoglobinemia and hemoglobinuria. Some destruction of intact C3b-coated RBCs may be mediated by splenic and liver sequestration via the C3b/C3bi receptors on macrophages. The DAT is positive usually only with anticomplement reagents, but exceptions occur. Sometimes, however, the drug-dependent antibody itself can be detected on the RBCs if the offending drug (or its metabolites) is included in all steps of the antiglobulin test, including washing.222
A variety of drugs induce the formation of autoantibodies reactive with autologous (or homologous) RBCs in the absence of the instigating drug (see Tables 54–2 and 54–3). The most studied drug in this category has been α-methyldopa, an antihypertensive agent that no longer is commonly used.73,74,75,76 Levodopa and several unrelated drugs also have been implicated.39,46,59,61,68,77,78,79,80,81,82,83,84,85,86 Patients with CLL treated with pentostatin,87 fludarabine,88 or cladribine90 are particularly predisposed to autoimmune hemolysis, which usually is severe and sometimes fatal.
Positive DAT reactions (with anti-IgG reagents) in patients taking α-methyldopa vary in frequency from 8 to 36 percent. Patients taking higher doses of the drug develop positive reactions with greater frequency.73,75,76 A lag period of 3 to 6 months exists between the start of therapy and development of a positive antiglobulin test. The delay is not shortened when the drug is administered to patients who previously had positive antiglobulin tests while taking α-methyldopa.75
In contrast to the frequent observation of positive antiglobulin reactions, less than 1 percent of patients taking α-methyldopa exhibit hemolytic anemia.74 Development of hemolytic anemia does not depend on drug dose. The hemolysis usually is mild to moderate and occurs chiefly by splenic sequestration of IgG-coated RBCs. α-Methyldopa has been proposed to suppress splenic macrophage function in some patients, and normal survival of antibody-coated RBCs in such patients may be related, in part, to this effect of the drug.223
The DAT reaction usually is positive only for IgG.11 Occasionally, weak anticomplement reactions also are encountered.11
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


