INTRODUCTION
SUMMARY
This chapter presents a current appraisal of our understanding of aging followed by a more detailed description of age-associated changes in hematopoiesis and their clinical consequences. Those who are older than the age of 75 years comprise a rapidly growing segment of the population. Marrow, like other organs, undergoes characteristic changes with advancing age, and many of these changes are evident by standard examination. For example, within the marrow space, hematopoietic cells occupy approximately one-half the volume at mid-life, with adipose tissue making up the difference. Yet, in the absence of disease, blood counts are generally maintained within a range established as normal for younger individuals. This is possible because hematopoietic stem cells increase in number with age and are of sufficient functional capacity to respond to homeostatic signals. Older people are more likely to have chronic diseases that may produce additional stress on marrow reserve. Anemia, for example, is present in just over 10 percent of community-dwelling individuals older than age 65 years; for those residing in nursing homes, the prevalence is closer to 50 percent. One distinction between anemias in older people compared with younger people is that for approximately one-third of older anemic patients, a specific cause for the anemia cannot be determined. This “unexplained anemia” is likely the result of multiple factors, including inappropriately low erythropoietin response, inflammatory cytokines, androgen deficiency, and, in some persons, incipient myelodysplasia. Platelet and neutrophil changes with age have been incompletely characterized but are likely to be subtle and of little clinical consequence. There is a well-characterized, age-associated involution of the thymus gland that precedes the histologic changes within the marrow, and marrow-derived T- and B-cell precursors are affected. Older people have fewer naïve, reactive T cells and an increase in relatively inert memory T cells. Thus, the capacity to react to new antigenic challenges is reduced and there is an increased susceptibility to certain infections and vaccines. Also evident are deficient regulatory functions, which may explain the observed increase in autoantibody, paraproteins, and inflammatory cytokines in those of advanced age. In the absence of disease, however, these alterations are of little consequence. In the presence of chronic debilitating disease, they are likely to become more pronounced and as such, contribute to an exaggerated decline in overall function. Similar conclusions can be drawn regarding dysregulated inflammatory pathways and coagulation. In balance, advancing age is associated with a procoagulant profile that may be of clinical importance in the presence of underlying atherosclerotic vascular disease.
Acronyms and Abbreviations:
EPESE, Established Populations for the Epidemiological Study of the Elderly; HSCs, hematopoietic stem cells; IADL, independent activities of daily living; IL, interleukin; NHANES III, National Health and Nutrition Examination Survey III; PAI-1, plasminogen activating inhibitor-I; TNF, tumor necrosis factor; UA, unexplained anemia; WHO, World Health Organization.
A PRIMER ON AGING
The world’s population is aging at an unprecedented rate and the implications for health care delivery are profound.1,2,3,4 Over the next several decades, the percentage of the population older than age 65 years will nearly double.5 In anticipation, an increased research effort is being made to better understand the basic biology of aging and the mechanisms whereby individuals become susceptible to disease.6,7
A central dogma of gerontology is that aging is not a disease. Yet, intrinsic biologic aging is the major risk factor for virtually all major diseases of developed societies, including cancer, diabetes, atherosclerotic cardiovascular and cerebrovascular disease, diabetes, neurodegenerative diseases (e.g., Alzheimer and Parkinson), osteoporosis and infection. Examination of the mechanisms by which biologic aging contributes to the pathogenesis of these diseases has now become recognized in the mainstream of scientific inquiry over a broad range of disciplines.7 However, from a biogerontologist’s perspective, there remain certain features of aging that transcend the more discipline-focused investigations.
One of these common features of aging is heterogeneity. For example, for any measureable variable, the range of values among normal older individuals is much wider than the range of normal among younger individuals.8,9 This variation is particularly relevant for hematologists consulted to examine older, but otherwise healthy, individuals with laboratory values outside the “normal range.”
Although functional declines that accompany normal aging have been well characterized,10,11 in general these are not of sufficient magnitude to account for symptoms or be mistaken for disease. For example, that kidney function declines with age is well recognized,12,13 and, in fact, has proven to be a useful biologic marker of aging. Yet, clinical consequences of this change in renal function, in the absence of a disease or the exposure to an exogenous nephrotoxic agent, do not occur commonly. Similarly, the marrow changes with age. Marrow stem cells increase in number and proliferative capacity, yet the in vitro proliferative potential of progenitor cells is less.14,15,16 Although clinically significant cytopenias do not occur in the absence of disease, mild to moderate anemia that has not been fully characterized occurs with increasing frequency, especially in the frail elderly.17,18,19,20 Furthermore, in frail individuals even a mild reduction in hemoglobin level is associated with untoward clinical outcomes.19,21,22
Certain immune functions decrease with age,23,24,25 but these may be of only marginal clinical significance. For example, whether the laboratory-observed declines in immune function contribute to a heightened susceptibility to infection is a subject of debate, but data support an association of age-associated qualitative change in lymphocyte function and susceptibility to reactivation of tuberculosis26,27 or herpes zoster28,29 and diminished response to influenza vaccine.30,31,32,33 There is compelling evidence that a profound decline in immunity is causally related to certain malignancies,34 but there remains debate over whether the more modest decline associated with normal aging is sufficient to account for the observed increased rate of cancer in the elderly.35,36 In fact, there is some evidence that cancer incidence is even lower in frail elderly,37 a population known to be functionally immunodeficient.38 Similarly, development of autoantibodies appear with increasing frequency with advancing age, but these are considered markers of an acquired humoral immune dysregulation, and with regard to autoantibody, not of clinical importance.39 In contrast, essential monoclonal gammopathy, a clonal B lymphocyte expansion that increases in frequency with age and that stabilizes at a clone size that does not impair normal immunoglobulin synthesis or inhibit hematopoiesis, does have a probability of undergoing clonal evolution to a B-cell malignancy, such as myeloma, lymphoma, or monoclonal light-chain amyloidosis at a rate of 1 percent per year (Chap. 106).40
Providing a rational, unifying explanation for the aging process has been the subject of a great number of theoretical expositions. Yet, no single proposal suffices to account for the complexities observed (Table 9–1).
That genetic controls are involved seems obvious when one considers that lifespan is highly species-specific. For example, mice generally live approximately 30 months and humans approximately 90 years. However, the aging phenomenon is not necessarily a direct consequence of primary DNA sequence. For example, mice and bats have 0.25 percent difference in their primary DNA sequence, but bats live for 25 years, 10 times longer than mice. Thus, regulation of gene expression seems likely to be the major source of species longevity differences.
Gerontologists have long been intrigued by the concept of accelerated aging and by examining those rare individuals who are so affected. From work with invertebrate models a number of genes have been identified that associate with longevity. Yet, the identification and functional analysis of analogous genes in humans remains elusive. With regard to genetic examples of accelerated aging, two syndromes have been well characterized: Hutchison-Guilford syndrome (early-onset progeria) and Werner syndrome (adult-onset progeria).41,42 Although neither these nor other progeria syndromes manifest a complete phenotype of advanced age, the identification of the genes responsible for these particular syndromes is beginning to pay dividends by providing clues to the molecular mechanisms involved in the aging process. For example, Werner syndrome is now defined by mutations in a single gene on chromosome 8 that encodes a protein containing a helicase-like domain.43,44 The activity of the Werner protein helps to maintain telomere structure and homology-dependent recombination.45 Similarly, a mutation in the lamin A (LMNA) gene localized to chromosome 1 has been causally related to the Hutchison-Guilford syndrome.46 The product of the mutated LMNA gene (termed progerin) accumulates, producing a variety of nuclear distortions of which telomere dysfunction and associated replicative senescence are notable.47
Examination of aging in yeast has also been informative with regard to the genetic controls of aging. These single-cell organisms follow the replicative limits of mammalian cells and it has been observed that “life span” is related to silencing large chromosomal regions. Mutations in these silencing genes lead to increased longevity.48
Thus, if there are certain genes that regulate normal aging, or at least are associated with the development of an aged phenotype, it stands to reason that acquired mutations of those genes might influence the rate of aging. Over the years several theories have been proposed that relate to this supposition. In general, they hypothesize a random or stochastic accumulation of damage, either to DNA or protein that leads eventually to dysfunctional cells, cell death and subsequent organ dysfunction, and ultimately death. Prominent among these is the somatic mutation theory,49 which predicts that genetic damage from background radiation, for example, accumulates and produces mutations that ultimately result in functional decline. A variety of refinements have been suggested to this theory, invoking the importance of mutational interactions,50 transposable elements,51 and changes in DNA methylation status.52
A related hypothesis is Burnet’s intrinsic mutagenesis theory,53 which proposes that spontaneous or endogenous mutations occur at different rates in different species and that this accounts for the variability observed in life span. Closely related to this notion is the DNA repair theory.54 Initially, there was great excitement about this idea as it was found that long-lived animals had demonstrably more active DNA repair mechanisms than shorter-lived species.54 However, longitudinal studies within a species have not revealed a consistent decline in repair mechanisms with age. This, of course, does not rule out the possibility that repair of certain specific and critical DNA lesions is altered with advancing age. We now understand that there are multiple DNA repair mechanisms, including base excision repair, transcription-coupled repair, and even mitochondrial DNA repair mechanisms. Disorders involving one or a subset of repair mechanisms could lead to accumulation of DNA damage and dysfunction.
In yet another intrinsic/stochastic model, Orgel55 proposed the error catastrophe theory in which he suggested that random errors in protein synthesis occur and when the proteins involved are those responsible for DNA or RNA synthesis, there is resultant DNA damage and the consequences thereof to daughter cells. Although this model has appeal, there has been no reported evidence for impaired or inaccurate protein synthesis machinery with advancing age. However, a candidate protein that may eventually be shown to be so affected is telomerase. This critical enzyme is necessary for maintaining telomere length and cell replicative potential. In vitro cellular senescence is associated with diminished telomerase activity,56 but whether this relates to aging of the organism as a whole remains controversial.57
Evidence that exogenous factors are involved in the acquisition of age-associated damage to DNA and protein is derived from a number of observations, many of which are circumstantial or correlative, but nonetheless provocative. It now appears that the accumulation of abnormal protein within senescent cells, as predicted by the error catastrophe theory, actually reflects posttranslational events, such as oxidation or glycation, and resultant crosslinking. There is theoretic appeal to the concept that key proteins, such as collagen or other extracellular matrix proteins, and DNA become dysfunctional with age as a consequence of the impairment produced by these crosslinks.58,59,60
One mechanism producing crosslinks is called glycation, the nonenzymatic reaction of glucose with the amino groups of proteins. Presumably, glycation would occur more readily in the presence of higher serum levels of glucose, and, thus, this theory fits well with the observed, age-associated dysregulation of glucose metabolism and prevalent hyperglycemia in geriatric populations. Of course, these findings also point out the theory’s deficiency as a unifying mechanism, as there is no question that individuals with well-maintained glucose levels throughout their life span will still be subject to the acquired changes typical of aging.
Another mechanism held responsible for crosslinking is the damage produced by free radicals, which forms the basis of the free radical hypothesis initially promoted by Harman.61,62 This theory offers that aging is the result of DNA and protein damage (e.g., mutagenesis or crosslinking) by atoms or molecules that contain unpaired electrons (free radicals). These highly reactive species are produced as byproducts of a variety of metabolic processes and are normally inhibited by intrinsic cellular antioxidant defense mechanisms. Nitrate-based free radicals are also generated by in vivo processes and another set of nitrogen free-radical scavenging mechanisms are in place. If free radical generation increases with age, or the defense mechanisms that scavenge free radicals (e.g., glutathione) or repair free radical damage decline, the accumulated free radical damage may account for altered DNA and protein function. Evidence to support this widely held notion is incomplete. It is known that free radical generation in mammals correlates inversely with longevity63 and, similarly, the level of free radical inhibiting enzymes, such as superoxide dismutase were higher in those species with longer life spans.63 However, efforts at enhancing antioxidant mechanisms with dietary vitamin E have resulted in only a modest enhancement of median survival in mice and no effect on maximum life span.64,65,66
Much attention has been focused on mitochondrial function in the context of free radical damage because the bulk of oxidative metabolism and the production of reactive oxygen species occur in these organelles. Although mitochondrial DNA codes for antioxidant enzymes in addition to enzymes involved in energy production, it is currently believed that energy production declines with age as a result of mitochondrial DNA damage by those reactive products. Indeed, mitochondrial damage increases with age in experimental models,67,68,69 and the shortened survival of knockout mice deficient in mitochondrial antioxidant enzymes has supported the potential importance of this mechanism.70
The most compelling data to date in support of the free radical hypothesis come from experiments in which transgenic Drosophila producing enhanced levels of superoxide dismutase and catalase had a maximum survival 33 percent greater than controls.26 Furthermore, it is known that flies produce high levels of free radicals associated with their impressive metabolic requirements, and that survival is enhanced dramatically when the ability to fly is experimentally hindered.65 However, the generalizability of these findings has been questioned. It has been noted that transgenic mice overexpressing free radical scavenging enzymes have produced very modest effects on life span.71 Thus, the conclusion that augmentation of free radical scavenging mechanisms increases longevity in mammalian species is not established.
From a different perspective, very good evidence implicates a nonrandom, perhaps genetically regulated endogenous mechanism involved in aging. For example, the neuroendocrine theory suggests that the decrements in neuronal and associated hormonal function are central to aging. It has been suggested that age-associated decline of hypothalamic–pituitary–adrenal axis function results in a physiologic cascade leading, ultimately, to the “frail” phenotype. This hypothesis is appealing because it is well established that this neuroendocrine axis regulates much of development and also the involution of ovarian and testicular function. Furthermore, age-associated declines in growth hormone and related factors,72 dehydroepiandrosterone,73 and secondary sex steroids74 are implicated in age-associated impairments, including a reduction in lean body mass and bone density. Furthermore, pharmacologic reconstitution using these or related hormones has met with some success at reversing age-associated functional decline.75,76
Similarly, it has been argued that involution of the thymus gland and subsequent decline in immune function discussed below is a key regulator of aging.77 The argument is based upon the observation that the decline in immune function occurs in all mammalian species, but occurs later in those with longer survival.78 Furthermore, dietary restriction is associated with maintained thymic mass and measurable immune function as well as prolonged survival, suggesting an association of a decline in immunity with primary aging processes.
The possibility is highlighted by the observation that differences in maximum survival of different mouse strains has been associated with specific alleles in the major histocompatibility complex, which, in turn, code for immunologic determinants.79 This hypothesis, although not without its appeal, is not widely accepted as a major explanation for aging. Perhaps this relates to the fact that biologic aging is a universal phenomenon and certain features are held in common, even in organisms with primitive or no immune function. (The same could also be said for the neuroendocrine theory.) It is obvious that the immune system is of great importance in minimizing the chance of early death, particularly from infectious diseases. However, immunologic reconstitution of middle aged or old animals has not been shown to prolong survival.80
From the perspective of those who study aging, an important distinction is made between median (life expectancy) and maximum life span. Over the past century, a dramatic increase in median survival has been mostly attributable to modern sanitation and refrigeration, as well as public health measures, including vaccination and antibiotics.81 Early deaths have been diminished and more individuals are reaching old age. In the United States today, expected survival from birth is approximately 80 years.82 Median survival is what concerns public health officials and healthcare providers. In contrast, maximum survival is the focus of those gerontologists interested in the biology of aging and longevity.
The oldest human being alive today is approximately 120 years old. It is intriguing that the oldest age limit has remained stable, unchanged by the public health initiatives mentioned above. In the laboratory, limits on age have been established for a variety of species. Drosophila, free of predators, disease or fly swatters, can live 30 days, whereas C57BL/6 mice in a laboratory environment and allowed to eat a healthy diet ad libitum, may survive 40 months. Unlike health-related interventions in humans, certain experimental interventions in lower species are associated with a prolongation of maximum survival. In Drosophila, for example, transgenic offspring producing extra copies of the free radical scavenging enzymes superoxide dismutase and catalase survive approximately 33 percent longer than controls.83 In nutritional intervention studies involving lower species, controlled restriction of dietary intake (dietary restriction) has become a common experimental paradigm exploited in the investigation of primary processes of aging and maximum survival.84,85
Dietary restriction typically involves a reduction of 30 to 40 percent in caloric intake with careful attention to the provision of adequate amounts of essential nutrients. It is associated with both a delay in the acquisition of age-related diseases (including cancer) and a reduction in the rate of achieving certain established biomarkers of aging (i.e., a retardation in primary aging). The critical questions remain: What is the mechanism of the effect of dietary restriction, and will it be applicable to higher species? With regard to the latter, there are now comprehensive and interactive studies within the United States in which dietary restriction is being examined in nonhuman primates86,87 and human studies are also underway.88,89 Although it appears that the calorie restricted monkeys in these studies are assuming a more youthful phenotype in a variety of physiologic measures,86,90,91 it remains too early to predict whether maximum survival will be affected.
After a finite number of divisions, normal somatic cells invariably enter a state of irreversibly arrested growth, a process termed replicative senescence.92 In fact, it has been proposed that escape from the regulators of senescence is what oncologists term malignant transformation. However, the role of replicative senescence as an explanation of organismal aging remains the subject of vigorous debate (for review, see references 93 and 94). The controversy relates, in part, to the fact that certain organisms (e.g., Drosophila, Cunninghamella elegans) undergo an aging process, yet all of their adult cells are postreplicative.
What is clear is that the loss of proliferative capacity of human cells in culture is intrinsic to the cells and not dependent on environmental factors or even culture conditions.92 Unless transformation occurs, cells age with each successive division. The number of divisions turns out to be more important than the actual amount of time passed. Thus, cells held in a quiescent state for months, when allowed back into a proliferative environment, will continue approximately the same number of divisions as those that were allowed to proliferate without a quiescent period.95 The question remains whether this in vitro phenomenon is relevant to animal aging.95 Although when various species are compared, replicative potential is directly and significantly related to life span,96 within an organism there is great variability in proliferative capacity from tissue to tissue and organ to organ. As such, age-associated changes in the marrow or gut might relate to replicative senescence, whereas in muscle or brain other processes most certainly are involved. But, added to this heterogeneity within an individual, is that fact that certain commonly employed models of aging (e.g., Drosophila, C. elegans) undergo an aging process despite a long held belief that all of their adult cells were post replicative.94,97,98 However, this notion has been countered by the demonstration of multipotent intestinal stem cells within the midgut near the intestinal basement membrane of Drosophila.99 Unlike intestinal stem cells in vertebrates that interact with stromal cells within a niche, analogous cells within Drosophila reside on the surface of the basement membrane and interact directly with daughter cells. Nevertheless, the presence of such cells has rekindled an interest in Drosophila as a model for stem cell biology, cancer, and whole-animal aging.100,101
A feature of cellular senescence is diminished proliferative capacity. In fact, it is now understood that genes considered tumor suppressors (e.g., p53, RB [retinoblastoma gene]) prevent cancer by inducing programmed cell death (apoptosis), particularly in cells at risk for neoplastic transformation. Alternatively, they can prevent potential cancer cells from proliferating by inducing permanent withdrawal from the cell cycle (cellular senescence). Although little is known about how cells choose between apoptotic and senescence responses, both are crucial for suppressing cancer102,103 and both are highly relevant to functional decline and longevity.104
There are a multitude of oncogenic stimuli that may result in either cancerous transformation or cellular senescence.105,106 Epigenetic changes within chromatin, histones, or nucleic acids may be caused by pharmacologic agents or altered expression of proteins.107,108,109 Such changes can alter the expression of protooncogenes or tumor-suppressor genes and are a frequent occurrence among malignant tumors. Thus the senescence response aborts the uncontrolled proliferative response an assortment of potentially oncogenic stimuli.
Although diverse stimuli can induce a senescence response, they appear to converge on one or both of the two pathways that establish and maintain the senescence growth arrest. These pathways are governed by the gatekeeper tumor-suppressor proteins p53 and pRB.104,110,111 Furthermore, the senescence response to dysfunctional telomeres requires the integrity of the p53 pathway.112,113 Overexpression of the RAS gene may also trigger a p53-dependent damage response by producing high levels of reactive oxygen species.113,114,115 However, oncogenic RAS can also induce p16, an activator of the pRB pathways, which provides a second barrier to the proliferation of potentially oncogenic cells. There is an emerging consensus that senescence occurs through one pathway or the other, with the p53 pathway mediating senescence primarily as a result of telomere dysfunction and DNA damage and p16/pRB pathway–mediating senescence primarily as a result of oncogenes, chromatin disruption, and various stresses.
A more speculative, but potentially important consequence of cellular senescence may be its impact on stem cells.116 Embryonic stem cells, whether human or rodent, express a high level of telomerase and thus are considered resistant to replicative senescence.117,118 However, mammalian adult stem cells or progenitor cells do not proliferate indefinitely.119,120,121,122 The ability of stem cells to undergo senescence and apoptosis is likely to be an important mechanism for preventing cancer.123,124
AGING AND HEMATOPOIESIS
Aging is a universal phenomenon that affects all normal cells, tissues, organ systems, and organisms. Accordingly, the marrow undergoes changes with age. Age-related hematologic changes are reflected by a decline in marrow cellularity, an increased risk of clonal myeloid neoplams125 and anemia,17,126,127,128,129,130 and a decline in adaptive immunity.131,132,133,134
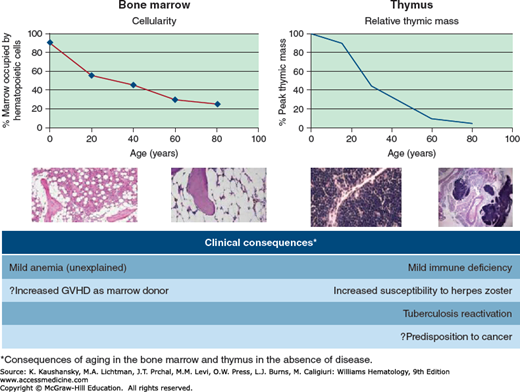
The percentage of marrow space occupied by the hematopoietic tissue declines from 90 percent at birth to a level of approximately 50 percent at age 30 years and 30 percent at age 70 years.135,136 A similar change occurs in the thymus, where involution begins at an earlier age and is reflected anatomically by a reduction in lymphoid mass with an increase in fat137 and functionally by a steady decrease in the production of naïve T cells.80,138 Fat infiltration into the marrow and thymus results in a diminished volume of hematopoietic tissue.
Although age-related change in the marrow is well described, the exact mechanisms that regulate these changes remains speculative.139 For example, it remains unclear whether the age-associated expansion of marrow fat is a cause or an effect of aging and whether the changes seen in marrow and histologically similar changes within the thymus are intrinsically related. Because of the intricate association of hematologic and immune functions and these common histologic patterns of change with age, both changes in blood and innate immunity are discussed below in the sections on Blood Cell Changes with Age and Aging and Immunity.
The ontogeny of hematopoietic stem cells is the focus of much attention.140,141 In fetal development the manufacture of blood cells occurs in several organs, but after birth this function is subsumed by the marrow.142,143,144,145,146,147,148 The process of embryonic and fetal hematopoiesis is described in Chap. 7. Hematopoietic cells appear in the medullary cavities of bone around 14 weeks of gestation,149 and by birth the marrow is the primary site of hematopoiesis.
Unlike the commonly held notion that stem cell compartments diminish either in number or function with age ultimately resulting in an inability to meet homeostatic demands, age-related hematopoietic stem cell (HSC) changes appear to be an exception, at least for murine species in which this question has been most directly addressed.150,151 Early work demonstrated that marrow serially transplanted could reconstitute hematopoietic function for an estimated 15 to 20 life spans.152 Furthermore, the capacity for old marrow to reconstitute proved superior to that of young.153 Subsequently, a number of investigators using a variety of techniques have concluded that HSC frequency in old mice and humans is approximately 2 to 10 times greater than in the young.16,150,151,154,155,156 Some evidence suggests that the intrinsic function of HSCs changes somewhat with age, most notably in a shift in lineage potential from lymphoid to myeloid development. This may contribute to an observed relative increase in neutrophils and decrease in lymphocytes in the blood of older people.157
There is an intrinsic change in HSCs with age, most notably resulting in a shift in lineage potential from lymphoid to myeloid development. This may contribute to a relative increase in neutrophils and decrease in lymphocytes in the blood of older persons.157 As HSCs age, they accumulate genotypic (mutational) and phenotypic alterations. Indeed, human stem-progenitor cells from healthy volunteers were found to accumulate 13 exonic (private) mutations per year of age.157a Current opinion is that such changes are responsible for the development of immune senescence and that such changes are responsible for the development of immune senescence, as well as the increased occurrence of age-associated diseases such as myelodysplasia and leukemia. Thus, the process of “immunosenescence,” as it affects the innate and adaptive immune system, may result from HSC aging. For example, an age-related decrease in the provision of B-cell precursors may be the result of HSC aging.148
The most apparent change seen in the marrow with aging is decreased cellularity (Fig. 9–1).135 Under normal circumstances, the marrow is the only site of hematopoiesis. Foci of extramedullary hematopoiesis may occur in the liver, spleen, or lymph nodes in pathologic states, but they are not of functional consequence. Until puberty the entire skeleton remains hematopoietically active, but by age 18 years only the vertebrae, ribs, sternum, skull, pelvis, proximal epiphyseal regions of humerus and femur remain active sites of blood production, with other medullary sites infiltrated with fatty tissue. By age 40 years, the marrow in sternum, ribs, pelvis and vertebrae is composed of equal amounts of hematopoietic tissue and fat and cellularity declines gradually thereafter. By age 65 years, marrow cellularity is estimated to be approximately 30 percent,135,136 with a corresponding increase in marrow fat. Age-associated imbalanced bone remodeling and osteoporosis results in decreased trabecular bone which itself may contribute to diminished hematopoiesis.158 The presence of fat correlates with the occurrence and severity of osteoporosis, both of which are evident with aging.159 Several age-related qualitative changes have been identified in hematopoietic cells, including skewed X-chromosome inactivation, telomere shortening,160,161,162 accumulation of mitochondrial DNA mutations,163,164 and micronuclei formation,165 any of which could result in cellular dysfunction. Furthermore, growth hormone production declines with age, and this, too, is linked with deposition of fat within the marrow.166 Administration of growth hormone to old rats reduces marrow fat and increases hematopoietic tissue.167
Figure 9–1.
Aging of marrow and thymus. Marrow cellularity declines from birth in a manner comparably to thymic mass. This is reflected histologically by the increased presence of fat. The clinical consequences of these age-associated changes, in the absence of disease, are a mild anemia and immune deficiency. The latter is reflected by an increased predisposition to certain infections (e.g., herpes zoster or reactivation of latent tuberculosis) and possibly to the increased predisposition to cancer. GVHD, graft-versus-host disease.
Anemia is a significant health problem in the elderly because of a high prevalence and significant associated morbidity, including reduced quality of life, clinical depression, falls, functional impairment, slower walking speed, reduced grip strength, loss of mobility, worsening comorbidities, and mortality.168,169
In older men and women, anemia defined using the World Health Organization (WHO) criteria of hemoglobin levels less than 13 g/dL for men and 12 g/dL for women170 is associated with an increase in mortality.171,172,173,174,175,176 It has been pointed out that the WHO criteria do not take into account inherent ethnic variations, particularly with respect to Americans of African descent who have lower levels of hemoglobin without significant adverse outcomes.177,178 In a study that analyzed 1018 Americans of African descent and 1583 Americans of European descent adults aged 71 to 82 years, anemia defined by the WHO criteria was associated with increased mortality in those of European descent but not those of African descent.177,178 The reasons for these ethnic differences are undefined. However, the difference is one of degree. In general, the impact of anemia on functional status and mortality in Americans of African descent becomes apparent at hemoglobin levels approximately 1 g/dL lower than in whites. The issue of establishing criteria for the diagnosis of anemia is relevant in the context of age, as well. Older women, for example, have better physical performance and function at hemoglobin values between 13 and 15 g/dL than at between 12.0 and 12.9 g/dL,179 suggesting perhaps that the cut off level of 12 g/dL is too low. Nevertheless, the WHO definition remains the standard used in most current epidemiologic surveys and many clinical laboratories.
In the third National Health and Nutrition Examination Survey (NHANES III) database, a nationally representative sample of community-dwelling persons and determined age- and sex-specific prevalence rates of anemia in the total U.S. population,127
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


