GUILLAIN-BARRÉ SYNDROME
TONY M. MCGRATH
Since the decline in cases of poliomyelitis, the Guillain-Barré syndrome (GBS) has emerged as the most frequent cause of acute flaccid paralysis. The spectrum of GBS includes classical acute inflammatory demyelinating polyneuropathy (AIDP), acute motor axonal neuropathy (AMAN), acute motor sensory axonal neuropathy (AMSAN), and variants such as Fisher syndrome (1,2). AIDP, the most common form in the Western Hemisphere, is characterized by rapidly progressive, usually ascending, and symmetric paralysis of the extremities, variable involvement of bulbar, facial, and ocular muscles, and loss of deep tendon reflexes (3). Although the disorder is often preceded by a bacterial or viral illness, no evidence presently exists to indicate that GBS is caused by direct infection of nervous tissue. Current research implicates an autoimmune mechanism.
HISTORY
Landry is credited with the first modern description of a disorder similar to GBS. In 1859, he reported a patient with paresthesias and subjective weakness. Unlike present definitions of GBS, objective weakness did not occur until 1 month after symptom onset but was then severe and rapidly progressive, leading to death 8 days later (4). Although Osler’s account in 1892 (5) of acute febrile polyneuritis was remarkably similar to the illness we now term GBS, the development of the lumbar puncture in the late nineteenth century permitted a more precise characterization of the disorder and its differentiation from poliomyelitis and other neuropathies (6). In 1916, Guillain et al. (7) detailed a clinical syndrome in two individuals characterized by motor weakness, paresthesias, areflexia with preservation of cutaneous reflexes, and increased cerebrospinal fluid (CSF) albumin without pleocytosis (albuminocytologic dissociation). Both recovered fully. Similar descriptions soon followed, but controversy arose regarding diagnostic criteria.
In 1976, following an apparent increase in GBS associated with the swine flu vaccine, the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) was directed by the Centers for Disease Control and Prevention (CDC) and the National Institute of Allergy and Infectious Diseases to develop clear diagnostic criteria for GBS to assist in epidemiologic studies. These criteria (NINCDS criteria) were reported in 1978 (3) and, though intentionally somewhat restrictive, are now generally accepted. The clinical section of this chapter contains a summary of these criteria.
EPIDEMIOLOGY
Epidemiologic studies of GBS are inherently troublesome (8). The major difficulty is the lack of definitive diagnostic markers for GBS. Because diagnosis must rely on clinical criteria, incidence may be over- or underestimated if criteria are too broad or stringent or if atypical variants are counted or excluded. Also, the results of studies employing dissimilar criteria may not be comparable. A dilemma arises in regard to whether persons with an underlying condition known to produce a neuropathy, such as diabetes mellitus, should be included when one is attempting a survey of a pure neuropathy such as GBS. Moreover, GBS displays a wide range of clinical severity. Because many epidemiologic studies rely on hospital-based reporting, mild cases not requiring hospitalization could be lost. Despite these difficulties, numerous epidemiologic investigations of GBS have been performed. Ten of these are summarized in Table 20.1.
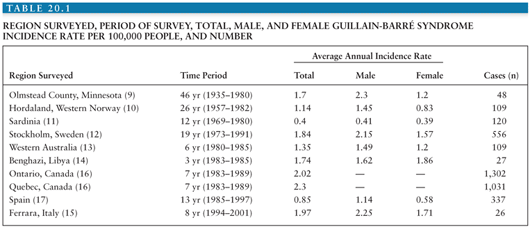
Incidence rates in studies using NINCDS criteria (9–14,15) range from 0.4 to 2.0 per 100,000 persons. In the population-based Olmstead County survey (9), 15% of GBS cases were mild and did not require hospitalization. Therefore, hospital-based studies (10,11,13,14) may underestimate the true rate by as much as 15%. Nevertheless, incidence rates are similar among studies conducted in different decades, ethnic groups, races, and geographic areas. These findings are consistent with the hypothesis that the triggering agents responsible for GBS are widespread and numerous, and susceptibility is similar among populations.
Most studies of GBS have revealed higher incidence among men (9–11,13) and no significant seasonal differences (9,13,16). Incidence increases with advancing age beyond 40 years, commonly peaking in the seventh and eighth decades (10,11,13,14,16,17). Autumn clustering of GBS following Campylobacter jejuni infection has occurred (18,19).
Four epidemiologic surveys have examined the incidence of GBS in children (20–23). Two (21,23) used NINCDS criteria, and two (20,22) used criteria outlined in Table 20.2 (24). Although children with clinical variants of GBS were identified, they were not included in the results. Annual incidence rates of GBS in children were lower (0.4 to 1.1 per 100,000 children) than rates obtained in the general population (see Table 20.1). No difference in frequency between boys or girls occurred in California (20) and Finland (22), whereas male-to-female ratios of 1.5:1 were observed in Paraguay (21) and Taiwan (23). Although a seasonal preponderance was not apparent in three studies (20,22,23), 76% of affected children in Paraguay (21) had onset of symptoms during the summer months.

When childhood GBS in California (20) was analyzed by age at onset, frequency was significantly greater in 2-year-old children than among other age groups. This finding may reflect the higher incidence of triggering infections in this age group. In Paraguay (21), a similar increase in children younger than 4 years of age was seen, but in Finland (22) and Taiwan (23), age distribution was evenly dispersed.
Antecedent Events
Clinically apparent antecedent infections occur within 28 days of onset in approximately 50% of persons with GBS (9–14,16, 25,26). Minor upper respiratory symptoms account for about 60% of the infections, followed by gastrointestinal ailments and nonspecific febrile illnesses. Elderly persons with GBS experience a similar incidence of antecedent infections (27,28). However, they may be less likely to complain of preceding gastrointestinal symptoms (27). As many as 70% of children with GBS experience an infectious disease within 28 days of neurologic symptom onset, with nonspecific upper respiratory illnesses being responsible in nearly 80% (20–23). Yet, gastrointestinal prodromic symptoms may vary. A recent study found that vomiting was more common in children with AIDP compared with AMAN (28.1% vs. 6.5%), and diarrhea was more common in AMAN compared with AIDP (32.6% vs. 12.5%) (19). Persons with severe GBS resulting in tetraparesis and prolonged mechanical ventilation are generally no more likely than mildly or moderately affected patients to have an antecedent infection (29–31).
Neurologic symptoms usually begin within 2 weeks after the infectious illness (9,13,16,26,27,30,31) but may occur more acutely (31). Shorter periods between the prodromal illness and neurologic symptoms seem to follow antecedent upper respiratory infections, but a longer delay may occur in patients with preceding gastrointestinal symptoms (32,33). In the majority, an infectious agent responsible for the prodromal illness is never identified.
Many viruses and bacteria (Table 20.3) have been implicated as antecedent triggering factors in GBS (16,21,28,32–36), although many of the proposed causal agents and GBS may simply represent a chance association. Winer et al. (26) tested the proposition in serologic studies on 100 persons with GBS and age- and sex-matched controls. Evidence of cytomegalovirus (CMV) and C. jejuni infection was significantly more frequent in GBS patients (11% and 14%, respectively) than among controls. Although positive Epstein-Barr virus (EBV), parvovirus B19, and Mycoplasma pneumoniae serologies were identified in GBS, controls demonstrated similar results. Hankey (13) obtained acute and convalescent serum from 62 persons with GBS and found evidence of recent viral infection in 50%. Parainfluenza virus type 3, CMV, and adenovirus titers were identified in five, six, and seven specimens, respectively, whereas EBV, herpes simplex, herpes zoster, parainfluenza type 1 and 2, measles, coxsackie, echovirus, coronavirus, and influenza A and B titers were each present in one or two samples. Infection with M. pneumoniae was encountered in five. Despite these various reports, convincing evidence of causality exists for only CMV, EBV, human immunodeficiency virus (HIV), herpes zoster, C. jejuni, and M. pneumoniae. Measles-associated GBS may occur, but only rarely. Although variola (smallpox) and vaccinia have been conclusively linked with GBS, the eradication of smallpox and the discontinuation of vaccinia immunization make any association with GBS of historic interest only. However, since the September 11, 2001 terrorist attack and the threat of bioterrorism, the expected increase in smallpox vaccinations could lead to an increase in vaccine-related GBS.
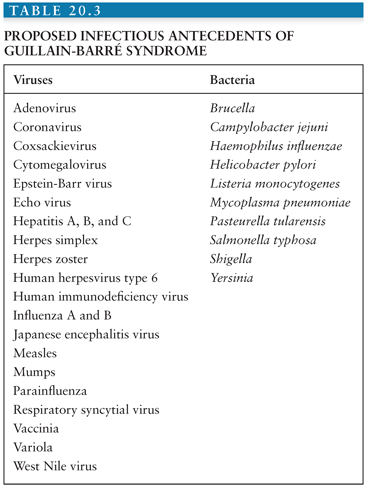
Dowling and Cook (36) detected immunoglobulin M (IgM) antibody directed against CMV in 15% of 220 persons with GBS. Those with CMV antibodies were primarily young women (average age, 25.6 years) in whom only 80% had a clinically apparent preceding infection. CMV-associated GBS appeared in three separate 10- to 16-week clusters over 3 years. Abnormal liver enzymes were present in 50% of the CMV seropositive cases. Evidence of recovery was associated with a fall in CMV IgM antibody titer. No correlation has been demonstrated between primary infection with CMV and severity or outcome of GBS (26,33). CMV-associated GBS has occurred following transplant of a CMV-infected heart in a CMV-negative recipient (37) and may occur with CMV reactivation after autologous bone marrow or organ transplantation (37,38).
EBV (like CMV, a herpesvirus) infection may precede neurologic symptoms in 2% to 8% of GBS (20,26,28). It may present as hepatitis or mononucleosis or, like CMV, may be clinically silent prior to GBS onset. Clinical or subclinical encephalopathy may complicate EBV-associated GBS (36). Antecedent primary herpes zoster infection may occur in 3% to 4% of children with GBS (20,23). GBS may also develop in association with reactivated herpes zoster infection (13,25,30). In contrast, recent herpes simplex virus infection is uncommonly identified in GBS (34). The most recently identified herpesvirus, human herpesvirus type 6 (HHV-6), has been proposed as an etiologic agent in GBS. Antibody against HHV-6 was demonstrated in a significantly greater number of GBS patients than among blood donors in Italy (35).
GBS has been described early in the course of HIV infection (39–41). In 1987, HIV was the most common infection associated with GBS at the University of Miami School of Medicine (41). In Zimbabwe, 55% of 29 consecutive persons with GBS were HIV seropositive (39). HIV infection did not produce a higher incidence of prodromal illness in GBS. In each seropositive individual, GBS was the initial manifestation of HIV infection. Generalized lymphadenopathy, coincident encephalopathy, and CSF pleocytosis were significantly more likely with HIV infection. Mean CSF white blood cell counts in HIV-associated GBS were 14/mm3 (range of 0 to 63 cells) (39). HIV seropositivity has not been associated with poor recovery following GBS (40,41). GBS in HIV-infected individuals is probably secondary to disordered immune regulation characteristic of the infection.
Several mosquito-borne infections have been reported with GBS. Japanese encephalitis virus (JEV) may be an important antecedent infection in endemic areas. JEV is the most frequently recognized cause of mosquito-borne encephalitis and typically occurs in epidemics in endemic areas. Ravi et al. (34) detected high titers of IgM antibody specific to JEV in serum and/or CSF in 62% (21 of 34) of consecutive cases in southern India. None had signs of encephalitis, and all satisfied NINCDS criteria (see later discussion) for GBS. Clinical course and outcome were similar in JEV-seropositive and -seronegative individuals. The JEV antigen was demonstrable in the CSF of one person, and virus was isolated from CSF in another. Pathologic examination in one autopsied case demonstrated discrete necrotic lesions at the cerebral gray–white matter junction and demyelination of ventral and dorsal nerve roots. West Nile virus (WNV) was reported in an individual with GBS during the 1999 outbreak in New York City (42). During the summer of 2002, six cases of acute flaccid paralysis syndrome associated with WNV were reported in Mississippi and Louisiana (43). The individuals, however, had asymmetric weakness and CSF pleocytosis, suggesting a polio-like syndrome. Falciparum malaria has also been reported with GBS (44,45). Sokrab et al. (45) reported a mortality rate of 40% in 10 individuals with acute falciparum malaria and GBS during a seasonal exacerbation.
In 1982, Rhodes and Tattersfield (46) reported the first case of GBS following C. jejuni enteritis. Subsequently, C. jejuni has been recognized as the most common bacterial antecedent infection in GBS. Serotyping of C. jejuni is accomplished by examination of heat-labile antigens (Lior method) and/or heat-stable antigens (Penner method) (47). Several Penner and Lior serotypes of C. jejuni have been isolated from GBS patients, but Penner serotype 19 and Lior serotype 11 predominate (48–58). These serotypes are uncommon in uncomplicated gastroenteritis (47). Winer et al. (26) and Boucquey et al. (33) found serologic evidence of recent C. jejuni infection in approximately 14% of GBS, and Mishu et al. (18) and Kaldor and Speed (59) reported C. jejuni infection in 37% of GBS. No history of preceding gastrointestinal symptoms has been reported in some GBS patients with serologic evidence of recent C. jejuni infection (33,59). The incidence of C. jejuni infection in the United States peaks from August through October (56), whereas the peak incidence for children in Mexico is from July through September (19). Statistically significant summer and autumn clustering of C. jejuni–associated GBS has occurred (18).
Stool cultures after onset of GBS with C. jejuni seropositivity are often negative (33,49,52,54,59), because C. jejuni excretion lasts an average of 16 days (60), and GBS symptoms may be delayed until 3 weeks after gastrointestinal symptoms. Appropriate antibiotic treatment of C. jejuni enteritis does not prevent GBS (46,49).
Although Boucquey et al. (33) and Enders et al. (54) reported no association between C. jejuni seropositivity and GBS severity or outcome, severe disease and poor prognosis in C. jejuni–associated GBS is well recognized (26,46,59,61). In one series of 21 C. jejuni–infected persons with GBS, 90% required mechanical ventilation (59). Severe involvement may occur because of a tendency of C. jejuni–associated GBS to damage axons rather than myelin (62). Of 11 children in China with AMAN, 90% demonstrated serologic evidence of recent C. jejuni infection (63). Conversely, Enders et al. (54) reported no greater electrophysiologic evidence of axonal involvement in C. jejuni–associated GBS, and although Vriesendorp et al. (61) demonstrated severe disease and poor recovery more commonly in C. jejuni–seropositive GBS, electrophysiologic evidence of primary axonal involvement was not apparent.
The mechanism by which C. jejuni infection triggers GBS is now better understood. Antigenic cross reactivity occurs between C. jejuni and components of peripheral nerves (PNs) (64,65). Host factors may determine whether infection with C. jejuni produces GBS. Although Hillert et al. (66) found no common human leukocyte antigen (HLA) class II antigens in GBS, and Winer et al. (67) demonstrated no association between HLA class I antigen and GBS and only a weak association between HLA class II antigen DR2 and severe GBS, Yuki et al. (52) reported HLA class I antigen B35 in seven Penner serotype 19 C. jejuni–associated cases of GBS. The authors concluded that the combination of HLA B35 antigen and Penner serotype 19 C. jejuni infection could precipitate an autoimmune response culminating in GBS. This is due to molecular mimicry between lipooligosaccharides (LOSs) of C. jejuni and gangliosides in PNs (68). The sialyltransferase gene (cst-II) has been identified to produce ganglioside-like LOS. The gene consists of 291 amino acids; the 51st determines its enzymatic activity. Cst-II (Thr51) produces GM1-like and GD1a-like LOSs, whereas Cst-II (Asn51) produces GT1a-like and GD1c-like LOSs which mimic GQ1b (69,70).
Evidence of M. pneumoniae infection has been confirmed in 1% to 5% of GBS (13,23,26,34). Currently, it is the second most commonly identified nonviral pathogen associated with GBS. An influenza-like syndrome may precede the onset of neurologic symptoms.
Mori et al. (71) reported elevated anti–Haemophilus influenzae antibodies in 6 of 46 (13%) Japanese patients with GBS but none of the 49 controls. Western blot analysis performed on the H. influenzae–positive patient’s immunoglobulin G (IgG) confirmed recognition both of lipopolysaccharides of H. influenzae and the ganglioside, GM1 (72).
In 1998, Chiba et al. (73) detected antibodies against Helicobacter pylori in the CSF from 4 of 7 patients with GBS. In 2002, Chiba et al. (74) examined CSF from 13 patients with GBS (8 AIDP, 4 AMAN, 1 unexcitable nerve conduction) and 8 control patients. Six of the AIDP patients had a specific IgG antibody to the VacA of H. pylori.
Rabies vaccine and the A/New Jersey/76 swine influenza vaccine (swine flu vaccine) have been reliably linked with GBS (32). Early rabies vaccine was made of desiccated brain and spinal cord of rabies-infected animals. Both acute demyelinating encephalomyelitis (ADEM) and GBS sometimes followed vaccination. A proliferative lymphocytic response and antibodies to myelin basic protein (MBP), a constituent of central and peripheral nervous system myelin, have been demonstrated in persons with rabies vaccine–associated GBS (75). An immune response against MBP has been proposed as the cause of ADEM and GBS in these individuals. The discontinuation of rabies vaccine prepared from infected animal brain and spinal cord has virtually eliminated rabies vaccine–associated ADEM and GBS.
In 1976 and 1977, an increase in GBS in the United States was temporally associated with the A/New Jersey/76 swine influenza vaccination campaign. The original CDC study demonstrated a statistically significant increase in the risk of developing GBS within 6 weeks of swine flu vaccine administration in persons 18 years of age and older (76). This study has been criticized because uniform GBS diagnostic criteria were not used, records were reviewed by nonphysician caseworkers, no standardized method of record assessment was used, and follow-up evaluations were not performed (77). Safrenek et al. (78) reviewed all GBS cases in Michigan and Minnesota from October 1, 1976 through January 31, 1977. Diagnostic criteria were developed, and a neurologist reviewed all medical records blindly. Although mistakes in the original CDC study were uncovered, these authors also demonstrated a significant increase in the relative risk of adults developing GBS within 6 weeks of swine flu vaccine. Four manufacturers produced the swine flu vaccine administered to the civilian population in 1976. All were associated with an increased risk of GBS (79). GBS has not been linked to previous or subsequent influenza vaccinations. Military personnel who received swine flu vaccine developed by the United States military in 1976 did not experience an increased frequency of GBS (80). The factor associated with the civilian vaccine that was responsible for the increased risk of GBS has never been determined; however, in 2008, Nachamkin et al. showed that the 1976 swine flu vaccine contained contaminating moieties which elicited anti-GM1 antibodies after inoculation into C3H/HeN mice. Vaccine samples from 11 unopened vaccine lots containing A/New Jersey/76 swine influenza antigen from three different manufactures were found to induce IgG and IgM antibodies to GM1, but no to C. jejuni, after immunization in mice. This response was also observed in both the 1991 to 1992 and 2004 to 2005 vaccines. No vaccines were found to contain bacterial DNA (81).
GBS has been reported following other vaccinations, but clear evidence of causality is lacking. Hankey (13) recorded recent vaccination in 5% of GBS, and of 100 individuals with GBS studied by Winer et al. (26), 6 had received a vaccination within 3 months of GBS onset. Ten vaccines were involved, and the rate of vaccination was similar in a control population. The latter study illustrates the importance of discovering the frequency of vaccination in the background population before assigning significance to a case of GBS following vaccination.
Several reports of GBS following vaccination against poliomyelitis have been recorded. In 1984, an outbreak of poliomyelitis in Finland resulted in an intensified vaccination program with oral poliovirus vaccine (OPV) (82). Subsequently, an unexpected rise in GBS occurred, precipitating epidemiologic surveys attempting to link OPV with GBS. Although an unexpectedly high rate of GBS occurred within 10 weeks following OPV administration, a small number of persons were involved, and an unexpectedly high rate of GBS also occurred in the weeks prior to the vaccination program.
Five percent to ten percent of GBS cases occur following intracranial, thoracic, abdominal, and orthopedic surgical procedures (6,13,26,29,30,33,37). This could be secondary to primary CMV infection following blood transfusion, although many reports exist of persons who did not receive transfusion. Other sources of iatrogenic infection associated with surgery or hospitalization could be responsible. Possibly, the surgery itself elicits a disordered immune response culminating in GBS.
GBS may also occur during pregnancy or the postpartum period (11,13,30,32). Unlike infants of mothers with myasthenia gravis, infants of mothers with GBS are unaffected. Whether the pregnancy or an associated infection, such as CMV, precipitates GBS is uncertain. Clinically and pathologically typical cases of GBS have also been associated with malignancy, especially Hodgkin and non-Hodgkin lymphoma (13,33,83). GBS is rarely associated with other autoimmune disorders (32).
PATHOLOGY
The pathologic findings in GBS are remarkably uniform irrespective of the antecedent illness or precipitating event. Lesions usually occur as discrete foci of abnormality and have been observed along the entire length of PNs. Areas of preferential involvement are the nerve roots, sites of ligamentous entrapment, and distal terminal branches (32). Kanda et al. (84) noted a relative absence of lesions in the initial few millimeters of anterior and dorsal nerve roots. This sparing of the transitional region between central and peripheral nervous systems (CNS and PNS) may be attributable to the marginal blood supply in this location. The pathologic process may involve autonomic, motor, and sensory systems simultaneously, but in individual cases, abnormalities may predominate in one. Areas of maximal pathologic change usually parallel clinical involvement, although this association is imperfect. Generally, motor involvement exceeds sensory changes, but in most patients, pathologic lesions do not preferentially or more severely involve the anterior roots or motor nerves (32,84–88). In a subset of GBS patients primarily manifesting motor dysfunction, relative sparing of dorsal roots and sensory nerves has been noted (63,85). The pathologic hallmark of GBS is a perivenular mononuclear cell infiltrate with segmental demyelination. In 1969, Asbury et al. described multifocal, perivenular mononuclear cell infiltration involving motor, sensory, autonomic, and cranial nerves (85). The invading lymphocytes and monocytes were derived from blood. Even in diffusely involved nerves, inflammatory cell concentration was greatest around blood vessels of the endoneurium, the connective tissue surrounding individual nerve fibers. Infiltration about vessels of the epineurium, the connective tissue sheath of the entire nerve, was present but less commonly observed.
Small and medium-sized lymphocytes were the predominant inflammatory cells recognized early; macrophages were the more prominent cell type in older lesions with considerable myelin destruction. Polymorphonuclear leukocytes were infrequently observed and then only in early, extensively affected areas, apparently in response to tissue necrosis. Plasma cells appeared in some chronic processes. Lesions of various ages were present within specimens from individual cases. Indeed, reparative processes were apparent in areas still presenting evidence of ongoing myelin breakdown (85).
Alterations of the myelin sheath occurred in the areas of inflammatory cell infiltration (85). Retraction at the node of Ranvier was seen in early lesions; more advanced changes included a complete dissolution of the myelin sheath. Myelin disintegration appears to evolve from the nodal region toward the centrally located Schwann cell. These areas of segmental demyelination were bounded by intervals of normal-appearing myelin. Active phagocytosis of myelin was evident. Axonal damage with resultant wallerian degeneration was present almost exclusively in areas with extensive inflammation. Asbury et al. (85) concluded that axonal degeneration was a secondary phenomenon induced by the severe inflammation and demyelination.
Schwann cell proliferation was present in lesions from patients surviving longer than 2 weeks. Although demonstrated in areas with segmental demyelination but preserved axons, Schwann cell proliferation was most common in nerves that had undergone wallerian degeneration. The proliferating Schwann cells were arranged in columns, forming a tube to guide the regenerating axons. Patients surviving several months with incomplete recovery of motor function demonstrated evidence of imperfect remyelination: myelin sheaths were thin and pale, internodes were shorter, and Schwann cell numbers were increased (85).
Subsequent examinations have supported many of the original findings of Asbury et al., but marked variability in the pathologic lesions of GBS is now commonly reported. These reports have described, even in early lesions, less conspicuous or absent lymphocytic infiltrate (63,84,86–91). Treatment with steroids or other immunomodulatory therapies may alter the degree of lymphocytic infiltration. Immunohistochemical analysis of biopsy material has demonstrated T-cell lineage in most of the invading lymphocytes with only a few B cells present (80,83). Although CD4+ helper/inducer and CD8+ cytotoxic/suppressor T cells are present, CD4+ cells may predominate in early lesions (88). Like Asbury et al. (85), Hanovar et al. (86) were unable to identify the origin of the larger mononuclear cells present in more advanced lesions.
Pathologic reports of GBS have confirmed the invariable presence of macrophages in areas of demyelination. Electron microscopic (EM) studies have demonstrated macrophages existing as two types (88–94). One type consists of actively phagocytic macrophages that occur in an extratubal location within the endoneurial space and in an intratubal location beneath the Schwann cell basement membrane. Focal lysis of the outermost myelin lamellae occurs in the areas contacted by the macrophage. The cytoplasmic projections of the macrophage then penetrate through the myelin gaps, insinuate themselves between the myelin lamellae, and peel away layers of myelin from the axon. These invading, actively phagocytic macrophages express major histocompatibility complex (MHC) class II antigens (86,88,95–98).
The manner in which macrophages accomplish myelinolysis is not understood. Arstila et al. (92) demonstrated increased acid phosphatase and acid proteinase activities in GBS lesions. These lysosomal enzymes may be derived from macrophages. However, the macrophage processes observed in GBS lesions are devoid of organelles, including lysosomes (90). The second type of macrophage, the foamy macrophage, is postphagocytic, contains myelin debris and lipids, and does not express MHC class II antigen (95). In GBS lesions, MHC class II antigen expression has also been observed on endothelial and Schwann cells (98).
Macrophages have also been observed within the axon cylinder (63). This location might explain some cases of axonal degeneration that apparently ensue in the absence of severe demyelination. McKhann et al. (63) reported pathologic findings on 10 autopsies from individuals with acute flaccid paralysis in China, a syndrome clinically indistinct from GBS. Although two of the patients had pathologic findings similar to those described by Asbury et al. (85), five were dominated by selective involvement of motor axons with absence of inflammation and little evidence of demyelination. Numerous macrophages were present in the areas of axons undergoing wallerian degeneration. The macrophages were located within both endoneurium and degenerating axons. The authors have designated this entity AMAN.
Immunoglobulin and activated complement deposits have been described in GBS nerve biopsy specimens. Nyland et al. (96) detected activated complement C3b receptor along nerves in GBS. Positive staining for C3 was observed along nerve fibers, and IgG and IgM were apparent in the region of the myelin sheath. Other investigators have failed to demonstrate immunoglobulin within the myelin sheath (88–90,94). Immunoglobulin staining of the endoneurium has occasionally been noted (89,90). Deposition of IgM on perineurium is a normal finding (89,90). Deposits of C3d were present in the myelin sheath of a single patient (94). In mice models of GBS, deposition of complement at the presynaptic motor nerve terminus has been reported (99,100,101). This in turn leads to a blockage of synaptic transmission at the neuromuscular junction (102).
Axonal degeneration in the absence of demyelination or inflammation has also been observed by Feasby et al. (88). Macrophages were not seen within the axon cylinder, and no evidence of inflammation or macrophage-associated demyelination was encountered. Axonal degeneration of motor and sensory nerves was severe. Electrophysiologic studies demonstrated inexcitability of motor nerves, a pattern seen primarily in axonal degeneration. The authors named this variant AMSAN. Prognosis for recovery is poor with axonal degeneration, whether it is a primary event or secondary to intense inflammation and demyelination.
PATHOGENESIS OF GUILLAIN-BARRÉ SYNDROME
The most compelling evidence for a T-cell–mediated mechanism in the pathogenesis of GBS is the finding of T-cell infiltration in affected nerves (32,85,87,88,90,91). Another indicator of T-cell involvement is the presence of lymphoblasts in blood during early GBS and a possible correlation between their number and disease severity (103). Increases in HLA-DR antigen and transferrin receptor, both present with T-cell activation, have been observed on the surface of T cells in GBS (104–108). Serum levels of interleukin-2 (IL-2), a T-cell growth factor, and serum-soluble IL-2 receptor (sIL-2R), shed from activated T cells, are increased in GBS (104–107). Levels are maximal at onset and decrease during recovery. Serum levels of IL-2 do not correlate with maximal functional disability. However, concentration of sIL-2R may predict severity (107). Activated T cells may mediate injury in GBS by (a) stimulation of B-cell proliferation and antibody production against myelin components, (b) macrophage recruitment, (c) activation of the complement system, or (d) direct cytotoxic damage of myelin or Schwann cells (97).
Consistent alterations in T-cell subpopulations have not been demonstrated (104,108–110). Dahle et al. (108) noted normal total lymphocyte counts in acute GBS but a decrease in the proportion of CD4+ helper/inducer T cells and an increase in the proportion of CD8+ cytotoxic/suppressor T cells. Within the CD4+ population, helper/inducer cells predominated over suppressor/inducer cells, known to be decreased in diseases of autoimmune mechanism. Winer et al. (110) demonstrated decreased numbers of CD8+ T cells in GBS patients with serologic evidence of recent C. jejuni infection and decreased numbers of CD4+ T cells in GBS patients with serologic evidence of recent CMV infection. Specific activation of T cells against myelin proteins has not been consistently observed with the sera from those with GBS. Two series (111,112) found no evidence of T-cell sensitivity to myelin P2 protein. Several reports have demonstrated T-cell clonal proliferation against proteins P2 and P0 and their peptides (113–115). However, similar results have been obtained in normal controls and controls with other neurologic diseases (113,115).
Macrophage involvement in GBS is evidenced by demonstration of macrophages in affected nerves in GBS pathologic specimens (32,85–88), electron microscopic observation of macrophage-mediated myelin stripping and phagocytosis (89–94), and indications of macrophage activation and their possible role as antigen presenters through the surface expression of MHC class II antigens. Neopterin is produced by macrophages in response to T-cell activation. Serum levels of neopterin are increased in GBS and fall as clinical involvement improves (116). In vitro, monocytes from GBS patients have demonstrated a greater ability to generate reactive oxygen species, indicative of cytokine activation, than monocytes from controls (104). Thus, available data indicate that macrophage activation, probably by activated T cells or their byproducts, occurs in GBS.
MHC class II marker expression is necessary for antigen presentation. Invading, actively phagocytic macrophages in GBS express MHC class II markers on their surface (86,88,89,95). Although MHC class II marker has been detected on the Schwann cell surface in GBS (98), these markers have also been observed on the Schwann cell surface in degenerative, toxic, and metabolic neuropathies (117). Tumor necrosis factor-α (TNF-α) is a cytokine elaborated by both activated T cells and macrophages. Increased serum levels of TNF-α occur in GBS (118). Although TNF-α may play a role in the immune-mediated injury in GBS, recombinant TNF-α alone will not produce myelin destruction in vitro (119).
The mechanism by which activated T cells and macrophages gain access to PN is not fully understood but apparently involves two adhesion molecules, E-selectin (ELAM-1) and intercellular adhesion molecule-1 (ICAM-1) (97). ELAM-1 is expressed on endothelial cells activated by IL-2, TNF-α, and endotoxin. It aids in the binding of lymphocytes, monocytes, eosinophils, and neutrophils to the blood vessel wall. Like IL-2 receptor, ELAM-1 enters the circulation and can be measured in serum. Elevated ELAM-1 serum levels have been observed acutely in GBS (120,121).
In recent years, the humoral immune system has been implicated in the pathogenesis of GBS because of (a) the demonstration of immunoglobulin and complement deposition in GBS nerve biopsy specimens (89,90,94,95), (b) the therapeutic effect of plasmapheresis and intravenous immunoglobulin (IVIG) (see later discussion), (c) the ability of GBS sera to cause demyelination of nerve in tissue culture and possibly in vivo, and (d) the demonstration of antibodies against PN constituents in GBS sera. Whole serum or IgM antibody from some patients with early GBS will produce complement-dependent demyelination of PN tissue cultures (122–124). Electron microscopic studies have disclosed damage to cultured Schwann cells and vesicular myelin degeneration after exposure to GBS sera in the absence of leukocytes, lymphocytes, or macrophages (122). A lesser degree of in vitro demyelination occurs in PN tissue culture after exposure to control sera, GBS patients during recovery, or persons with other neurologic diseases (123).
A humoral mechanism in the pathogenesis of GBS has focused on the detection of antibodies to PN constituents, especially gangliosides and related glycolipids. Complement-fixing antibodies directed against PN myelin are demonstrable in the sera of some persons with early GBS (120,125). Anti–PN-myelin antibodies are highest at presentation, significantly increased over levels from normal and diseased controls, and decrease with improving clinical symptomatology (125). Production of anti–PN-myelin antibodies in GBS is predominantly extrathecal, although measurable amounts are present in CSF (126). In GBS, activated complement components are detectable in serum and CSF. Anti–PN-myelin antibodies peak prior to activated complement levels, providing evidence of an antibody-mediated complement attack on PN myelin (127,128).
Characterization of anti–PN-myelin antibodies has disclosed increased levels of circulating antibody to galactocerebroside (GalC) (110) and P2 protein (112). Antibody levels to these myelin constituents were low and, in the case of P2 protein, only marginally increased over those of control sera. Koski et al. (129) discovered anti–PN-neutral glycolipid antibodies in GBS sera. Antigenic cross reactivity was demonstrated between this PN neutral glycolipid and Forssman antigen, a component of some viral and bacterial cell membranes.
Gangliosides are sialylated glycosphingolipids located on mammalian cell plasma membranes. Ganglioside function is not known precisely, but gangliosides are implicated in cell differentiation, cell–cell interactions, and receptor functions such as binding of viruses, bacterial toxins, hormones, growth factors, and interferons (130). IgG and IgA antibodies to GM1, GD1b, and GQ1b have been shown to activate leukocytes, which may contribute to the pathogenesis of GBS (131). The major gangliosides of PN are GM3 and GD3, and the major ganglioside of PN myelin is LM1. GM1, although a major component of CNS myelin, is only a minor constituent of PN. GM1 has been localized to both nerve plasma membrane and the myelin sheath and appears to accumulate in the paranodal region (132). Antibody to gangliosides is present in low amounts in normal human plasma (133). In 1988, Ilyas et al. (134) detected significantly increased levels of antibodies to the gangliosides GD1a, GD1b, and GT1b and to two associated glycolipids in serum from patients with acute GBS compared to controls.
High IgG antibody titers against GQ1b, a minor ganglioside of PN that has been localized to the paranodal region of human ocular motility nerves, was detected in 90% of 41 persons with Fisher syndrome (FS) (135–137), a GBS clinical variant manifesting primarily as ophthalmoplegia and ataxia. Anti-GQ1b antibody decreases as clinical symptoms improve and may be absent in people with mild FS (135–137). High anti-GQ1b antibody titers have also been demonstrated in Bickerstaff brainstem encephalitis, a syndrome that bears some clinical resemblances to FS (138), and in typical GBS with prominent ophthalmoplegia (137). High-titer anti-GQ1b antibody is not usually detectable in typical GBS lacking ophthalmoplegia or in normal controls (137). The pathogenetic role of anti-GQ1b antibodies in FS is not known. Lipopolysaccharide of C. jejuni isolated from FS does cross-react with epitopes of anti-GQ1b antibody (56).
Subsequent to the original observation of high antiganglioside antibody titers in GBS (134), antiganglioside antibodies (AGA) have been detected in 10% to 60% of patients with GBS (61,97,139–145). The AGA titers may correlate with GBS severity and decrease with clinical improvement (143). Antibodies to many known gangliosides and unidentified glycolipid fractions have been demonstrated in association with GBS, but antibody to GM1 is most frequently cited, especially in cases with mainly motor symptoms (139,141–144,146). Both IgM and IgG antibodies to gangliosides are present, but IgM antibodies predominate early (142). The early presence of IgG or IgA antibodies to GM1 has been associated with severe, prolonged GBS (139) and may predict poor recovery (141,144,147–149). AGA have also been detected in individuals with hyperreflexia and impaired central motor conduction in association with acute paralysis, acute ataxia, or chronic paralysis (150).
Anti-GM1 antibodies have been reported in up to 58% of patients with GBS showing serologic evidence of preceding C. jejuni infection (139,149). Yuki et al. (62) described anti-GM1 antibody in association with C. jejuni infection in severe GBS with electrophysiologic evidence of primary axonal degeneration and poor recovery. Immunogenic cross-reactivity between GM1 and C. jejuni isolates from GBS patients has been described (53,57). GM1 antibodies recognize a carbohydrate sequence that is present in several gangliosides and on bacterial cell walls. GM1 gangliosides may also be important for nerve growth factor signaling. Tanaka et al. reported that GM1 antibodies interfered with the neurotrophic action of nerve growth factor and TrkA autophosphorylation signaling in PC12 cells, a sympathetic nerve cell line (151). Nakatani et al. reported GM1 antibodies of patients with AMAN inhibit the current action of Cav2.1 (P/Q-type) voltage-dependent calcium channel in Purkinje cells of rats (152).
Elevated titers to GM1 and GD1b occur in C. jejuni infection without GBS (153). However, research now shows that elevated antibodies to GM1, GD1a, GM1b, and GalNAcGD1a occur with higher frequency in AMAN, whereas elevated GQ1b, GT1a, GD3, and GD1b antibodies occur in the majority of FS (154). In 32 Japanese children with GBS, Nishimoto et al. (155) reported the frequency of IgG antibodies to gangliosides: GM1 (34%), GM1b (22%), GD1a (25%), and GalNAc-GD1a (13%). In addition, antibodies to ganglioside complexes (GSCs) are associated with severe GBS. Patients with GBS requiring mechanical ventilation may have antibodies to GD1a/GD1b and GD1b/GT1b complexes, and patients with FS may have antibodies to GQ1b/GM1 and GQ1b/GD1a GSC (156,157,158,159). GM1/GalNAc-GD1a GSC have also been reported in 10 patients with GBS (160). Other antibodies associated with GBS have been identified (Table 20.4) (65,162–164). Immunogenic cross reactivity between GalC and M. pneumoniae has also been described (165).

Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


