INTRODUCTION
SUMMARY
The introduction of next-generation sequencing platforms, coincident with genome-scale preparatory and analytical approaches and the completion of the Human Genome Reference, has ushered in the era of genomics. This chapter introduces the fundamentals of next-generation sequencing methods, provides an overview of the basics of data analysis, and explores the myriad applications developed to exploit the scale and throughput of next-generation sequencing toward questions of biomedical importance. Specifics of cancer genomics, complex disease genomics, and how they pertain to hematologic basic science and clinical practice are discussed, along with the modern-day realities of the consenting process.
Acronyms and Abbreviations:
AML, acute myeloid leukemia; ATAC-seq, uses the hyperactive Tn5 transposase to simultaneously fragment and add sequencing adaptors to accessible DNA; bp, base pair; ChIP-seq, chromatin immunoprecipitation sequencing; ddNTP, di-deoxynucleotide triphosphate; DNase-seq, uses DNase I to fragment DNA based on DNase I hypersensitive sites as a marker of chromatin accessibility; dNTP, deoxynucleotide triphosphate; FAIRE-seq, formalin crosslinking of DNA to proteins prior to random fragmentation; FFPE, formalin-fixed, paraffin embedded; FLT3-ITD, internal tandem duplications of FLT3 gene; Gb, gigabase, i.e., billion base pairs; GINA, The Genetic Information Nondiscrimination Act; GWAS, genome-wide association study; lncRNA, long noncoding RNA; indel, term for the insertion or the deletion of bases; MDS, myelodysplastic syndromes; miRNA, microRNA; MNase-seq, micrococcal nuclease (MNase) determines nucleosomal footprints and boundaries by pairing with NGS as a marker of chromatin accessibility; MRD, minimal residual disease; NGS, next-generation sequencing; PCR, polymerase chain reaction; RNA-seq, RNA sequencing; siRNA, short-interfering RNA; SNP, single nucleotide polymorphism; snoRNA, small nucleolar RNA; snRNA, small nuclear RNA; Tb, terabase, i.e., trillion base pairs; WGBS, whole-genome bisulfite sequencing; ZMW, zero-mode waveguide.
HISTORY OF GENOMICS: SANGER SEQUENCING
The scientific discipline known as genomics has dramatically changed since the publication of the Human Reference Genome in 2003, primarily as a result of the introduction and broad-based implementation of new sequencing technologies.1 Prior to the mid-2000s, Sanger sequencing was the predominant DNA sequencing approach, and was used to complete the sequencing of the first human reference genome. Frederick Sanger and his colleagues developed Sanger or “chain termination” sequencing in the late 1970s.2 In their original method, four reactions were used to accomplish chain termination by incorporating separate di-deoxynucleoside triphosphates (ddNTPs), each included with a mix of three unmodified deoxynucleoside triphosphates (dNTPs) and a fourth, radiolabeled dNTP. Each reaction consisted of the DNA template to be sequenced in a mixture containing a DNA primer, a DNA polymerase, a mixture of four dNTPs, and one of the four ddNTPs. Here, the chemistry of ddNTPs, which lack the 3′ hydroxyl group present in a native dNTP, resulted in chain termination when incorporated into a growing DNA chain, as DNA polymerase cannot add another nucleoside without the 3′ hydroxyl group present. With multiple rounds of primer elongation, the ddNTPs incorporate randomly in the newly synthesized strands according to the complementary nucleotides of the DNA template. By denaturing the newly synthesized strands from the DNA templates and resolving each of the four DNA fragment mixtures on separate lanes by gel electrophoresis, one could read out the sequence of the DNA template from the resulting autoradiograph. Significant improvements to the original Sanger sequencing protocol included the use of fluorescently labeled ddNTPs to allow for sequencing to occur in one reaction rather than four,3 improved thermally stable DNA polymerases that permitted temperature cycling (“cycled sequencing”), and the use of capillary electrophoresis rather than standard gel electrophoresis for automated separation matrix filling between samples.4,5,6,7 Modern Sanger capillary sequencers typically generate DNA sequencing reads in the range of 400 to 900 base pairs (bp). The main limitation of Sanger sequencing is that the sequencing reaction is decoupled from the electrophoretic separation and detection steps. To piece together the sequence for a large segment of DNA or entire genome, genomic DNA must be randomly fragmented and subcloned into a bacterial vector, with each cloned DNA isolated and sequenced. The resulting sequencing reads are assembled computationally to recreate larger fragments that recapitulate the starting DNA nucleotide sequence. This process is expensive, time-consuming, and laborious. However, with the availability of robotic DNA isolation and sequencing reactions, coupled with high-throughput capillary sequencers, the human genome, among the genomes of many other organisms, was decoded. Currently, Sanger sequencing is still in use to complete smaller scale sequencing projects and to validate findings from next-generation sequencing studies.
MODERN GENOMICS: NEXT-GENERATION SEQUENCING
The method for next-generation sequencing (NGS), or massively parallel digital sequencing, is distinct from Sanger sequencing in that the sequencing reactions alternate with cycles of signal detection to provide the data readout at a significantly accelerated scale.8,9 The use of NGS in the years after the completion of the Human Genome Project has greatly increased the use of genomics and has significantly impacted the pace of biomedical research.10 Although there are several different NGS platforms offered commercially, they are methodologically quite similar. Unlike Sanger sequencing, NGS does not require subcloning of DNA, propagation in a bacterial host, and isolation of individual templates prior to sequencing. Instead, DNA is randomly fragmented into a pool of small pieces (generally 100 to 500 bp) and then ligated with specific synthetic DNA linkers (or adaptors) at the fragment ends to generate a NGS “library.” The library fragments are subsequently amplified by a process that isolates individual library fragments to a specific location prior to amplification. In general, this in situ amplification occurs on a covalently modified surface (a bead or flat silicon surface) with complementary linkers covalently attached to it, using a specific dilution of library fragments as input. In this step, the individual library fragment amplification permits sufficient signal output for detection during the sequencing steps that follow. Because each sequencing read derived from an amplified library fragment originates from that single unique fragment, NGS data are digital in nature. This fact underlies an important concept for digital sequencing methods: the number of specific sequencing reads generated is directly proportional to the amount of input nucleic acid, accurately reflecting amplified regions of a genome, for example. However, as the generation of libraries and the amplification of fragments involve polymerase chain reaction (PCR) amplification, inaccuracies can result via amplification biases or from PCR enzyme substitution errors in which the wrong base is incorporated during amplification.
Currently, there are two commercially available NGS platforms in common use. One uses an approach called sequencing by synthesis that occurs in the microfluidic channels of a silicon-derived “flow cell” device (Fig. 11–1).11 Here, enzymatic amplification of library fragments on the flow cell surface results in hundreds of millions of DNA clusters, and the sequencing of each cluster occurs in parallel with all of the other clusters by a stepwise series of events. Solexa marketed the first commercially available sequencer using this technology in 2006, and was acquired by Illumina in 2007. Illumina offers a variety of different sequencing machines with varying run times (from hours to days), sequencing capacities (from 25 million reads to nearly 3 billion reads per flow cell), and overall output (from approximately 0.5 gigabase (Gb) to greater than 1.5 terabase (Tb) of sequenced bases per run).
Figure 11–1.
Illumina library construction and sequencing process. Panel A represents the library construction process whereby high-molecular-weight genomic DNA is fragmented, ligated with adaptors, and amplified on a solid support prior to annealing of adaptor-complementary primers. Panel B represents the stepwise sequencing process whereby reagents are introduced to extend the primed fragments, the incorporated fluorescent nucleotides are detected, the 3′ end is deblocked, and the fluorescent groups on the incorporated nucleotides removed prior to the next stepwise sequencing-by-synthesis series. (Reproduced with permission from Mardis, ER: Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif) 2013;6:287-303.)
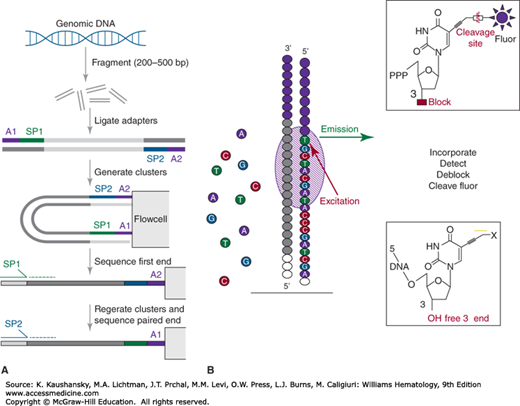
As in Sanger sequencing, the sequencing-by-synthesis steps begin with annealing sequencing primers complementary to the adaptors to the amplified library fragments on the flow cell surface. Then, a solution containing DNA polymerases and fluorescently-labeled, chemically modified dNTPs are added to the flow cell to begin an incorporation step. The DNA polymerases incorporate the complementary dNTP onto the 3′ ends of the primed fragments in each cluster. Each incorporation reaction is terminated after a dNTP is added, because of a blocking group at the 3′ position. After a cycle of dNTP incorporation on the flow cell, a laser-based detection system scans the flow cell surfaces to excite the incorporated fluorescent groups and to collect the unique light emission of each of the four fluorescently labeled dNTPs. Chemical deblocking steps follow to (1) remove the fluorophore by cleavage (the fluorescently-labeled dNTPs are known as “reversible dye terminators”) and (2) unblock the 3′ hydroxyl group to permit the next cycle of incorporation, detection and blocking.
Unlike Sanger sequencing, Illumina’s sequencing-by-synthesis method generates relatively short read lengths, typically 100 to 300 bp. The limitations on read length are primarily a signal-to-noise issue, where increasing numbers of steps in the sequencing-by-synthesis approach produces increasing noise at each step that competes with true signal detection. Hence, the data quality of Illumina reads tends to decrease with increasing step numbers. Illumina error rates are low, in the 0.1 to 0.3 percent range, and the predominant error type is base substitution.12 Ultimately, a complex, repetitive genome such as the human genome cannot be assembled from 300-bp read lengths, so algorithms were developed to align reads to the reference genome as a first step toward data interpretation.13 One approach by which Illumina has improved read mapping is by enabling paired-end sequencing that permits the sequence read off first one end and then the other of each amplified fragment cluster on the flow cell. Paired end reads of this type physically are linked and defined by the fragment size, permitting their accurate placement onto the reference genome by alignment, and effectively permitting more reads to contribute to coverage from a given sequencer run (when compared to single-end reads).14 Furthermore, as described later, the expected read placement onto the reference genome, when not met, is a source of information used to interpret structural variation.
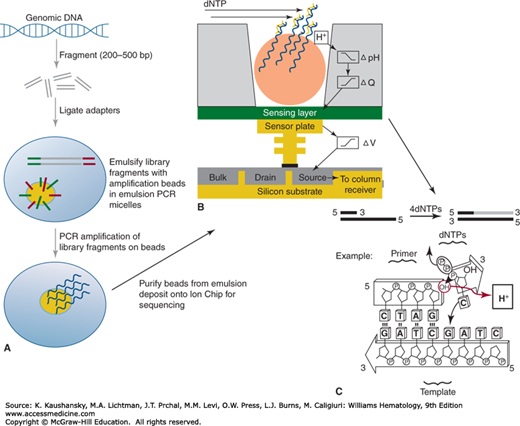
The second type of NGS platform in common use is the sequencing by pH sensing method that is marketed by Life Technologies (now a part of Thermo Fisher) as their Ion Torrent platform (Fig. 11–2). Life Technologies acquired Ion Torrent in 2010.15 The sequencing by pH-sensing method involves similar steps of library construction as described for sequencing by synthesis. However, the library DNA fragments are diluted and combined with (1) individual micron-scale beads that have covalently attached complementary adaptors on their surface and (2) PCR reagents, including DNA polymerase, into an emulsion PCR reaction. In emulsion PCR, one generates individual aqueous micelles that permit bead-based amplification of library fragments prior to sequencing. The emulsion PCR process generates beads carrying copies of identical DNA fragments suitable for sequencing. The DNA-coated beads are purified from the emulsion, enriched for those beads with amplified DNA on their surfaces, and then deposited into individual wells of a specifically constructed semiconductor plate, known as an Ion Chip. Sequencing primers (complementary to the adaptors) are annealed to the bead-amplified fragments, and then the sequencing process is initiated by the addition of DNA polymerase and flow of a single dNTP-containing buffer solution across the Ion Chip surface. The flow of the four nucleotides occurs in a stepwise fashion, with a detection step and an intervening wash. When a specific dNTP is incorporated into the elongating strands of DNA fragments on a specific bead, hydrogen ions are released, and a highly sensitive pH sensor built into the Ion Chip can read out the subsequent change in pH for the well containing that bead. If no dNTP is incorporated at that cycle, no change in pH is registered for that well. This approach follows for all wells containing beads on the Ion Chip, resulting in massively parallel sequencing. As with the Illumina technology, read lengths are short, in the 100 to 400 bp range. Unlike the Illumina platform that uses paired-end reads, Ion Torrent sequencing reads are single-end reads. The source of most sequencing errors generated by the Ion Torrent platform are insertion/deletion errors in stretches of identical bases on the template strand as a result of the difficulty of discerning the pH change ratio associated with incorporation of the same nucleotide above four consecutive identical nucleotides.16 Advantages of the Ion Torrent are that run times are very short (in the 2- to 7-hour range), and the cost per run is relatively inexpensive. The output, read length, run time, and cost vary by the Ion Chip type used (up to 2 Gb).
Figure 11–2.
Ion Torrent library construction and sequencing process. Panel A represents the specifics of the Ion Torrent library amplification process, which requires an emulsion PCR amplification on the surface of a bead with covalently attached adaptor-complementary primers, followed by emulsion breaking and bead addition to the Ion Chip for sequencing. The sequencing process, illustrated in panel B, flows sequential high-purity dNTP solutions across the chip surface for incorporation. Upon incorporation, there is a release of hydrogen ions that are detected by the pH-sensing capability of the chip, detected in panel C. (Used with permission from Thermo Fisher Scientific.)
There is one commercially available platform for single-molecule sequencing, the Pacific Biosciences RSII instrument.17 Single-molecule sequencing differs primarily from the previous platforms discussed in that no PCR amplification is required prior to data generation. This has obvious advantages in eliminating some sources of bias that result from the use of PCR, but has a disadvantage in that higher input amounts of DNA are typically required. The other major difference in the Pacific Biosciences approach is in the read length obtained, which ranges according to the template type but can exceed 50,000 bases with the input of very long molecules to the library construction.
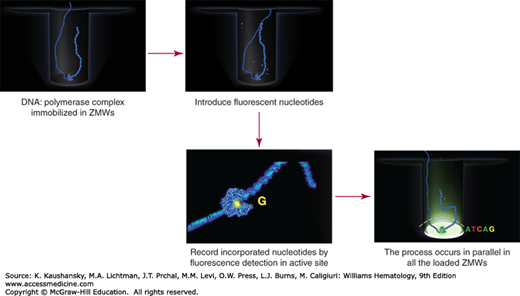
The Pacific Biosciences approach couples primed DNA library fragments with DNA polymerase molecules that are specifically engineered for the sequencing system (Fig. 11–3). These complexes are introduced to the surface of a SMRTCell, a nanofabricated sequencing device, which consists of 150,000 zero-mode waveguides (ZMWs). In effect, the loading of complexes aims to place one DNA polymerase/DNA template complex into each ZMW in preparation for sequencing. The ZMW is a nanofabricated pore that focuses the laser excitation and detection optics at the bottom of the ZMW where the DNA polymerase complex is bound, isolating the detection area to the active site of the polymerase. The sequencing process initiates with the introduction of fluorescent nucleotides and buffers, and is continuously monitored by the excitation/detection optics during the run time. As fluorescently tagged nucleotides sample into the active site, they can be detected with sufficient dwell time upon their incorporation into the synthesized strand. Because each fluorescent group is specific to the nucleotide identity, the sequence is read out based on the detected emission wavelength. The fluorescent group is attached to the phosphate portion of the nucleotide, so incorporation removes it by cleavage during the phosphodiester bond formation, and it diffuses out of the ZMW focus.
Figure 11–3.
Pacific Biosciences real-time sequencing and detection process. The primed library fragments are complexed to DNA polymerases and applied to the surface of a SMRTCell, where they locate into zero-mode waveguides (ZMWs). After providing fluorescently labeled nucleotides and buffer, the sequencing process is monitored by real time detection, whereby incorporated nucleotides are detected in the active site of each ZMW-isolated polymerase complex by the laser/detection optics of the instrument. Here, the fluorescence events are recorded for each active ZMW throughout a preset duration, resulting in the final sequencing read data for each single DNA molecule. (Used with permission from Pacific Biosciences.)
Single-molecule sequencing has, by definition, an inherently higher error rate as a consequence of the signal-to-noise ratio associated with detecting a single event in real time. The predominant error type in Pacific Biosciences sequencing reads is an insertion/deletion error that may be a result of inaccuracies in detecting (1) a nucleotide that had a longer than average dwell time but was not incorporated, (2) a single nucleotide that incorporated but was mistaken for two (or more) nucleotides, or (3) by errors in detecting multiple nucleotide incorporations into a homopolymer stretch. In spite of an approximate 15 percent error rate, the errors are essentially random, which means that oversampling (or “coverage”) of the sequence of interest can correct most errors, resulting in a cumulative error rate of around 0.1 percent following read assembly.18 The very long read lengths possible on this platform enable read assembly, rather than read alignment needed in short read platform data analysis. Assembly has obvious advantages in its ability to represent novel content in a genome and to provide long-range haplotyping information.
Emerging DNA sequencing technologies are being developed around the central concept of translocating DNA molecules through nanopores, which can be either biologic or nanofabricated pores.19 In nanopore sequencing, detection of the nucleotide sequences occurs during nanopore translocation events that are sensed by changes in electrical current that correlate to sequence, or by laser-based detection of incorporated fluorescent nucleotides. Nanopore sequencing, while still somewhat theoretical, may offer rapid sequencing with very long read lengths.
Initially, NGS platforms were used for whole-genome sequencing of organisms with relatively small genomes, such as bacteria or model organisms (Caenorhabditis elegans, Drosophila melanogaster, etc.), or for combining large numbers of PCR products into a single sequencing run. As the throughput per run improved, larger genomes, including human genomes, were studied, including the first cancer genome.20,21
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


