INTRODUCTION
SUMMARY
The term gene therapy describes treatment resulting from expression of a transferred gene (or transgene) in diseased or other cells by engineered vectors. Once within the cell, the transgene can direct synthesis of a therapeutic protein that can complement a genetic deficiency or confer upon the cell a desired phenotype or function. Many clinical trials have involved gene therapy for patients with various gene-deficient hematologic diseases, such as severe combined immunodeficiency, hemophilia, Wiskott-Aldrich syndrome, chronic granulomatous disease, aplastic anemia, hemoglobinopathies, HIV infection, and leukemia. Results from some clinical trials indicate that gene therapy can cure or improve many inherited or acquired hematologic disorders. This chapter reviews the basic principles of gene transfer and the results of selected preclinical and clinical studies.
Acronyms and Abbreviations
AAV, adeno-associated virus; ADA-SCID, adenosine deaminase deficiency severe combined immunodeficiency; ARSA, arylsulfatase A; BCNU, 1,3-bis-(2-chloroethyl)-1-nitrosourea; CAR, chimeric antigen receptor; CCR5, chemokine (C-C motif) receptor 5 gene; CGD, chronic granulomatous disease; CLL, chronic lymphocytic leukemia; CRISPR, clustered, regularly interspaced, short palindromic repeats; DSB, double-stranded break; FA, Fanconi anemia; FVIII, factor VIII; FIX, factor IX; GCV, ganciclovir; GVHD, graft-versus-host disease; Hgb, hemoglobin; HR, homologous recombination; HSC, hematopoietic stem cell; HSV, herpes simplex virus; HSV-TK, herpes simplex virus thymidine kinase; iCasp9, inducible caspase 9 protein; IL2RG, interleukin-2 receptor gene; LTRs, the long terminal repeats; MGMT, O6-methylguanine-DNA methyltransferase; MLD, metachromatic leukodystrophy; SIN, self-inactivating; siRNA, small interfering RNA; TALEN, transcription activator-like effector nuclease; TMZ, temozolomide; WAS, Wiskott-Aldrich syndrome; X-ALD, X-linked adrenoleukodystrophy; X-SCID, X-linked severe combined immunodeficiency; ZFN, zinc-finger nuclease.
DEFINITION AND HISTORY
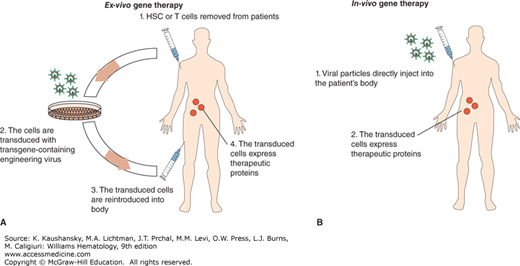
Gene therapy is a promising treatment for several inherited or acquired hematologic disorders. Gene therapy involves the introduction of a functional gene to replace a mutated gene or a therapeutic gene to provide a missing or defective protein to the organism. In some cases, the patient’s blood cells are removed and special, targeted cells such as hematopoietic stem cells (HSCs) are selected for engineering. The therapeutic genes are introduced into a vector and delivered into the targeted cells. These targeted, gene-modified cells are reinfused back into the patient. Because this method modifies cells outside the patient’s body, it is called ex vivo gene therapy (Fig. 29–1A). By contrast in vivo gene therapy describes the therapeutic gene-containing vectors being directly injected into the patient (Fig. 29–1B). In the in vivo case, the gene is expressed, producing a therapeutic protein for treatment. Theoretically, if the gene-modified cells are long-lived and able to expand inside the body, a single gene therapy can be sufficient to provide a lifelong therapeutic effect. Current gene therapy technologies have reached the point that many types of single-gene hematologic deficiency diseases can be permanently corrected, for example, X-linked severe combined immunodeficiency (X-SCID) and adenosine deaminase deficiency severe combined immunodeficiency (ADA-SCID).
Figure 29–1.
A. Ex vivo gene therapy involves 4 steps: 1. Obtain patient’s blood (HSCs or T cells); 2. transduce the cells in the laboratory by engineering the virus that contains a transgene; 3. infuse the transduced cells into the patient; and 4. the infused transduced cells produce therapeutic proteins. B. In vivo gene therapy is done by directly injecting viral particles into the patient.
Gene therapy has improved significantly in effectiveness since the mid-1980s when the first experiment using stem cell gene transfer was successful. Scientists initially encountered several serious obstacles. First, there was difficulty in delivering a modified gene into HSCs because of their lack of cell-surface receptors and their quiescent state.1 Second, early X-SCID gene therapy was interrupted because 20 percent of patients developed a T-cell type of leukemia within 3 to 6 years after therapy. This event was caused by gene therapy-related viral vector insertional mutagenesis; the vector contained powerful enhancer elements within its long terminal repeats (LTRs) that inserted close to, and activated the LMO2 protooncogene.2 Over 3 decades, these problems have been (for the most part) resolved. With an array of cytokine stimulation cocktails to improve HSC receptivity to engineering, and improved lentiviral vectors, the HSC transduction rate in humans can reach 80 to 100 percent.3,4 In addition, myeloablative conditioning regimens (e.g., busulfan, melphalan and 1,3-bis-[2-chloroethyl]-1-nitrosourea [BCNU; aka carmustine]) that decrease the number of endogenous stem cells prior to infusion of the engineered HSCs has proven to be an effective method to increase engraftment rates (average: 1 to 2 copies of gene marking/cell).3,4 High-level stem cell engraftment is a critical factor for the gene therapy of chronic granulomatous disease, X-linked adrenoleukodystrophy (X-ALD) and metachromatic leukodystrophy.3,4,5,6 Moreover, newer, safer self-inactivating (SIN) viral vectors have been developed in which viral LTR enhancers are completely removed. Using these newer vectors, no patient has developed therapy-related malignancy in several clinical trials, some of which have followed patients for as long as 8 years.3,4 Gene therapy is no longer a hypothetical form of therapy; some trials have achieved clinical correction for as long as 12 years. Gene therapy has, thus, undergone a renaissance. This chapter discusses two critical technical factors in gene therapy: targeted cells and delivered vectors; briefly reviews the gene therapy of several common hematopoietic genetic deficient diseases; and describes new strategies of in vivo selection and insertion site-targeted gene therapy.
GENE THERAPY TARGETED CELLS
HSCs are the cell of choice for many gene therapy applications for several reasons. First, many blood cell disorders originate at the stem cell level and, therefore, gene-corrected HSCs are the best candidate for replacement. Second, HSCs are a long-lived and self-renewing population and may reduce or eliminate the need for repeated administration of gene therapy. HSCs are readily obtained in the blood, marrow, or umbilical cord blood. They are also easily selected and manipulated in the laboratory and can be returned to patients relatively easily. Third, engineered HSCs are able to correct defects in all hematopoietic lineages. Fourth, HSCs migrate to several tissues in the body—primarily the marrow, but also the liver, spleen, and lymph nodes. These may be strategic locations for localized delivery of therapeutic agents for disorders unrelated to the hematopoietic system, such as for patients with liver diseases.
The use of hematopoietic progenitor or precursor cells for gene therapy theoretically would require repeat application. However, for some inherited and acquired diseases involving T cells (e.g., severe combined immunodeficiency [SCID] and AIDS), the use of T-cell precursors is capable of long-term correction of the T-cell deficiency. The underlying mechanism is still not clear, but some types of T cells, such as memory T cells, are long-lived. The memory T-cell compartment has three subsets: T-memory stem cells, central memory T cells, and effector memory T cells.7 Memory T cells and central memory T cells can expand to large numbers of effector T cells when activated. T-cell gene therapy is effective in some forms of cancer therapy. Long-term effects could be related to memory T stem cells. For example, T effector cells can directly eradicate malignant cells. However, cancer cells are often “invisible” to T cells. For this reason, modified T cells have been created by adding a chimeric antigen receptors (CARs) expressing gene, engineering the T cells to recognize cancer-specific antigens such as CD19 on chronic lymphocytic leukemia (CLL) cells.8,9 Once the CAR-modified T cells are transfused into the patient, they can attack and eradicate the targeted malignant cells. In clinical trials, anti-CD19 CAR T cells profoundly decreased the level of cancer cells in three patients with end-stage CLL.8,9
An advantage of using gene-modified T cells is that the procedure does not disturb the otherwise normal hematopoietic system. However, this is also a limitation of using T cells in gene therapy. For example, when using T cells to correct X-SCID, although T cells recover to near normal levels, the other affected cell types (e.g., B cells, natural killer [NK] cells) are not corrected. In this approach, the patient would require immunoglobulin replacement and the NK cell deficiency would persist. A study compared the outcomes of several clinical trials of HIV therapy comparing CD4+ T-cell treatment to HSC-based treatment. These trials found that overall, the results from HSCs were better than that of CD4+ T cells.10 One reason offered for this result is that without stimulation, T cells proliferate at a very low rate, estimated at approximately one division every 3.5 years for naïve T cells, and one division every 22 weeks for memory T cells.11
GENE THERAPY VECTORS
The majority of gene therapy studies thus far have employed viral vectors, because of their high efficiency of transgene delivery into the human nucleus. For a long-term effect, genome-integrating retroviral/lentiviral vectors have been employed. However, DNA integration causing critical gene mutagenesis has raised concern about long-term safety. This concern has led to the development of persisting but nonintegrating viral vectors.
Adenoviral vectors do not integrate into the genome, but are highly effective in gene therapy because of their ability to efficiently transduce both dividing and nondividing cells, and to persist relatively well in long-lived targeted cells. Adenoviral vectors also have capacity to hold large segments of DNA (e.g., 7.5 kbp); they are easily manipulated with recombinant DNA techniques and have the ability to produce high titers.12,13 However, adenoviral vector infection enlists a variety of humoral and cellular immune responses.14 Therefore, adenoviral vector therapy may result in acute toxicity and autoimmunity, and which clears transgene-expressing cells, reducing the efficacy of therapy.13 Of interest, this autoimmunity can be targeting to adenovirus-infected cancer cells, and thus used as an oncolytic agent.15
The adeno-associated viral vector (AAV) has the abilities to infect both nondividing and dividing cells and to persist without vector integration.16,17 Upon entering host cells, wild-type AAV DNA becomes episomal or integrates into the genome; in contrast, current, modified AAV vectors are designed to lose their integrating ability.18 AAV can carry a transgene up to 4.7 kbp. The AAV genome is single-stranded DNA and vector preparations are composed of a mixture of vector particles having one of the two strands of the virus. Upon transduction, the required annealing of the two strands delays gene expression. This limitation can be overcome by using a self-complementary design in which the two strands of the transgene are on a single hairpin genome in an inverted orientation, which allows quick assembly into a transcription unit following transduction.19
A feature of retroviral vectors is that they have the ability to stably incorporate their viral DNA into the host genome, which can result in long-term expression of the transgene. Most retroviral vectors are γ-retroviral or lentiviral vectors. Lentiviral vectors are usually HIV-1 based, or may also contain elements of simian immunodeficiency virus, and have at least three advantages over γ-retroviral vectors; first, the lentiviral vector preintegration complex is able to cross the nuclear membrane in host cells even in the absence of mitosis and, therefore, is able to transduce nondividing cells, such as HSCs.20 Second, the lentiviral vector preintegration complex is more stable and persists longer, which improves the likelihood of integration.20 Third, γ-retroviral vectors prefer to integrate near gene transcription start sites, such as the CpG islands and conserved noncoding sequences and conserved transcriptional factor binding sites, whereas lentiviral vectors are more evenly distributed, reducing the likelihood of driving the expression of a deleterious gene(s).22 Lentiviral vectors have thus emerged as an important advance for gene therapy in the hematologic disorders.
GENE THERAPY FOR HEMATOLOGIC DISEASES
X-SCID is a single-gene-deficient disease caused by mutations in the gene of the interleukin-2 receptor, γIL2RG (Chap. 80).23 Lack of functional γIL2RG results in a lack of T and NK cells and poorly functional B cells. X-SCID is a fatal disease, and without medical interventions, patients often die within the first 2 years of life.23 During 1999 to 2006, 20 X-SCID children were treated in two gene therapy trials.24,25 Because patients lacked an human leukocyte antigen (HLA)-identical donor, precluding stem cell transplantation as a curative therapy, all 20 patients were given a single γ-retroviral vector-mediated gene therapy in which a wild-type γIL2RG gene was delivered into patients’ T cells. From 5 to 12 years of observation after the gene transfer procedure, 17 of the 20 treated participants are alive and display nearly full correction of the T-cell deficiency by genetically modified T cells.24,25
Adenosine deaminase (ADA) deficiency is an autosomal-recessive inherited disorder caused by mutation of ADA gene on chromosome 20 (Chap. 80). ADA deficiency leads to inhibition of DNA synthesis, which is particularly toxic to lymphocytes because they are some of the most mitotically active cells. This condition ultimately causes SCID. ADA-SCID is almost always fatal by 2 years of age. Gene therapy with a normal ADA gene expressed in autologous HSC is a potentially curative treatment. However, early attempts at gene therapy did not shown any clinical benefit. In the late 1990s, when improved retroviral vector and preinfusion chemotherapy conditioning were instituted, success occurred. Since 2000, 40 patients have been treated in Italy, Great Britain, and the United States.26,27,28 In these studies HSCs were transduced with a γ-retroviral vector encoding the ADA enzyme. Low-intensity conditioning with either busulfan or melphalan was used to increase engraftment of stem cells. In some patients the corrected ADA gene can be detected in blood mononuclear cells for more than 9 years, and the ADA enzyme remains at a normal level.27
Wiskott-Aldrich syndrome (WAS) is the result of a mutation in the gene encoding the WAS protein (WASp).28 The gene is located on the short arm of the X chromosome and WAS is an X-linked recessive genetic disorder. WASP activates actin polymerization in almost all blood cells. Hereditary deficiency in WASP is associated with microthrombocytopenia, recurrent infections, eczema, high incidence of autoimmunity and hematopoietic malignancy (lymphoma or leukemia).29 In 2010, three patients in Italy were administered a lentiviral vector engineered to express WASp following busulfan conditioning. All three WAS patients showed excellent multilineage engraftment with an average of 0.4 to 0.9 correct gene copy per genome persisting to 30 months. Symptoms of WAS improved substantially. Pretreatment eczema resolved between 6 and 12 months after therapy. The frequency and severity of infections progressively decreased, and cytomegalovirus replication was controlled, allowing withdrawal of antiinfectious prophylaxis in two patients. Improvement of platelet count resulted in discontinuation of platelet transfusions.3
There are two major leukodystrophies, X-ALD and metachromatic leukodystrophy (MLD); both have been successfully treated with gene therapy.4,6 X-ALD is a severe genetic demyelinating disease caused by mutations in the ABCD1 gene on the X chromosome that encodes the adrenoleukodystrophy (ALD) protein, an adenosine triphosphate binding cassette transporter. ALD deficiency leads to the accumulation of very-long-chain fatty acids and progressive demyelination in the central nervous system. In 2009, two French children were reported to have autologous HSC gene therapy with a lentiviral vector encoding wild-type ABCD1.6
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


