INTRODUCTION
SUMMARY
Deficiency of either folate or cobalamin (vitamin B12) leads to macrocytic anemia with or without other cytopenias as a result of megaloblastic hematopoiesis, a manifestation of defective DNA synthesis. Folate in its tetrahydro form is a transporter of one-carbon fragments, which it can carry at any of three oxidation levels: methanol, formaldehyde, or formic acid. The oxidation levels of the folate-bound one-carbon fragments can be altered by oxidation and reduction reactions that require nicotinamide adenine dinucleotide phosphate in its oxidized (NADP) or reduced (NADPH) form. The primary source of the folate-bound one-carbon fragments is serine, which is converted to glycine as its terminal carbon is transferred to folate. The one-carbon fragments are used for biosynthesis of purines, thymidine, and methionine. During biosynthesis of purines and methionine, free folate is released in its tetrahydro form. During biosynthesis of thymidine, tetrahydrofolate is oxidized to the dihydro form and must again be fully reduced by dihydrofolate reductase to continue functioning in one-carbon metabolism. Methotrexate acts as an anticancer agent because it is an exceedingly powerful inhibitor of dihydrofolate reductase, thereby interdicting the generation of reduced folate.
In the cell, folates are conjugated by the addition of a chain of seven or eight glutamic acid residues. These residues enable the retention of folates in the cell. When folates are absorbed from the intestine, a process that occurs chiefly in the duodenum and proximal jejunum, all but one of the glutamates is removed by the enzyme glutamate carboxypeptidase II (folate hydrolase). Resulting monoglutamate forms are then taken up by one of two folate-specific transporters located on the apical brush border small bowel epithelium, the reduced folate carrier or the proton-coupled folate transporter. Blood folates are taken up by cells, mainly in the form of methyltetrahydrofolate monoglutamate. The newly absorbed folates are rapidly reglutamylated in the cell by the enzyme folyl-polyglutamyl synthase (FPGS). If glutamylation is prevented, the folates cannot be retained in the cell, resulting in an intracellular folate deficiency.
Cobalamin is required for two reactions: intramitochondrial conversion of methylmalonyl coenzyme A (CoA), a product of catabolism of branched-chain amino acids, and ketogenic amino acids to succinyl CoA, a Krebs cycle intermediate and cytosolic conversion of homocysteine to methionine, a reaction in which the methyl group of methyltetrahydrofolate is donated to the sulfur atom of homocysteine. In cobalamin deficiency, methyltetrahydrofolate accumulates because, for practical purposes, donation of the methyl group to homocysteine is the only method of generating free tetrahydrofolate from methyltetrahydrofolate. Free tetrahydrofolate is an excellent substrate for FPGS; methyltetrahydrofolate is a poor substrate. Consequently, much of the methyltetrahydrofolate taken up by a cobalamin-deficient cell leaks out of the cell before it can be polyglutamylated. The megaloblastic anemia of cobalamin deficiency results from an intracellular folate deficiency that arises because of the cell’s limited ability to polyglutamylate methyltetrahydrofolate.
Absorption of cobalamin is a highly complex process. Upon arriving in the stomach, cobalamin is taken up by haptocorrin (HC) binder (also called R binder or cobalophilin), a glycoprotein found in virtually all secretions. When the cobalamin HC complex enters the duodenum, the HC is digested and the cobalamin is released into the intestinal lumen, where it is taken up by intrinsic factor, a glycoprotein secreted by the gastric parietal cells. The cobalamin-intrinsic factor complex is absorbed by cells in the ileum through receptor-mediated endocytosis, involving cubilin and other proteins. The cobalamin is released within lysosomes and transported to the blood where it circulates bound to transcobalamin (TC), which delivers its cargo of cobalamin to cells throughout the body. Folate (vitamin B9) and cobalamin (vitamin B12) play key roles in the metabolic machinery of proliferating cells.
Megaloblastic anemia most commonly results from folate or cobalamin (vitamin B12) deficiency. Folate deficiency often was nutritional in origin. It may be seen in alcoholics, the elderly, the poor, but also is seen in patients on hyperalimentation, with hemolytic anemia, or hemodialysis. In the many countries that now practice folic acid fortification of the diet, such as the United States and Canada, the prevalence of folate deficiency has been dramatically reduced and nutritional folate deficiency has been virtually eliminated. In pregnancy, even a mild folate deficiency may be associated with defects in neural tube closure in the fetus, so pregnant women should always receive folate supplements. The incidence of neural tube defects has fallen considerably in North America since the introduction of folic acid fortification. Diagnosis of folate deficiency is based on measurements of folate in serum, which furnishes information about the current level of folate, and in red cells, which provide data on aggregate folate status over the preceding period during which those red cells were produced. Nutritional folate deficiency is treated with folic acid by mouth.
Folate deficiency as a result of malabsorption occurs in tropical and nontropical sprue. Folate deficiency as a result of tropical sprue is treated with folate supplements and antibiotics. In nontropical sprue, the treatment is folate plus a gluten-free diet.
The most common cause of clinically apparent cobalamin deficiency is pernicious anemia (PA), a condition in which the portion of gastric mucosa that contains the parietal cells is destroyed through an autoimmune mechanism. The parietal cells secrete intrinsic factor, which is essential for physiologic cobalamin absorption. Without intrinsic factor, a state of cobalamin deficiency develops over the course of years. Cobalamin deficiency leads not only to megaloblastic anemia but also to a demyelinating disease that manifests itself as peripheral neuropathy, spastic paralysis with ataxia (so-called combined system disease of the spinal cord), dementia, psychosis, or some combination of these features. “Subtle” cobalamin deficiency, which may manifest as neurologic symptoms without anemia, appears to be relatively widespread among the elderly. The incidence of gastric cancer is increased by a factor of two to three in patients with PA. Other causes of cobalamin deficiency are gastric resection; stasis of the small intestinal contents as a result of blind loops, strictures, or hypomotility; and disease or resection of the terminal ileum, the site of vitamin B12–intrinsic factor complex absorption. Individuals on a vegan diet can become cobalamin deficient. Cobalamin deficiency is diagnosed by measuring the level of either total or TC-bound vitamin in the blood or by measuring serum methylmalonic acid, which accumulates in the bloodstream in patients with cobalamin deficiency. The cause of cobalamin deficiency was determined by the Schilling test, a measure of cobalamin absorption, but the test is obsolete and no replacement is currently available. In patients with nutritional megaloblastic anemia, folate or cobalamin deficiency as the cause of the anemia must be determined. If a patient with cobalamin deficiency is treated with folic acid, the anemia may be corrected but the neurologic abnormalities persist, progress or may be aggravated. Patients with cobalamin deficiency usually are treated with parenteral cobalamin but large doses of oral cobalamin may be used.
Megaloblastic anemia can develop as an acute disorder with rapid development of leukopenia and/or thrombocytopenia. Nitrous oxide anesthesia or abuse is responsible for some cases of acute megaloblastic anemia. The anemia is rarely also seen in patients with a marginal folate status in intensive care units or severe hemolytic anemia through increased folate demand for augmented erythropoiesis. The condition resembles an immune cytopenia but can be ruled out by examining the marrow, which exhibits a floridly megaloblastic picture.
Other causes of megaloblastic anemia include drugs (e.g., hydroxyurea, nucleoside analogues) and certain inborn errors of metabolism. Of the inherited conditions, TC deficiency is singled out because it causes a severe megaloblastic anemia in infants who respond completely to high-dose cobalamin. Irreversible neurologic complications supervene if the deficiency is not detected in time. Megaloblastic-like morphologic features of varying degree are seen in the myelodysplastic syndromes, and in acute leukemia of the erythroleukemia type. Megaloblastic anemia seen in association with refractory anemia with excess sideroblasts occasionally responds to very high doses of pyridoxine.
Acronyms and Abbreviations:
AdoCbl, adenosylcobalamin; AICAR, 5-amino-4-imidazole carboxamide ribotide; ATPase, adenosine triphosphatase; AZT, azidothymidine; CnCbl, cyanocobalamin; CoA, coenzyme A; CUB, cubilin; CUBAM, the binary ileal cubilin receptor complex consisting of cubilin and amnionless; dTMP, deoxythymidine monophosphate; dU, deoxyuridine; dUMP, deoxyuridine monophosphate; FH4, tetrahydrofolate; FPGS, folylpoly-γ-glutamyl synthase; [3H]Thd, [3H]thymidine; HC, haptocorrin; HCl, hydrochloric acid; IM, intramuscular; LDH, lactate dehydrogenase; MCV, mean corpuscular volume; MeCbl, methylcobalamin; MRI, magnetic resonance imaging; MTHFR, methylenetetrahydrofolate reductase; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate (reduced form); N2O, nitrous oxide; OHCbl, hydroxocobalamin; PA, pernicious anemia; PteGlu, pteroylglutamic acid (folic acid); SAH, S-adenosylhomocysteine; SAMe, S-adenosylmethionine; TC, transcobalamin.
FOLATE
Folate and cobalamin (vitamin B12) play key roles in the metabolism of all cells, particularly proliferating cells.
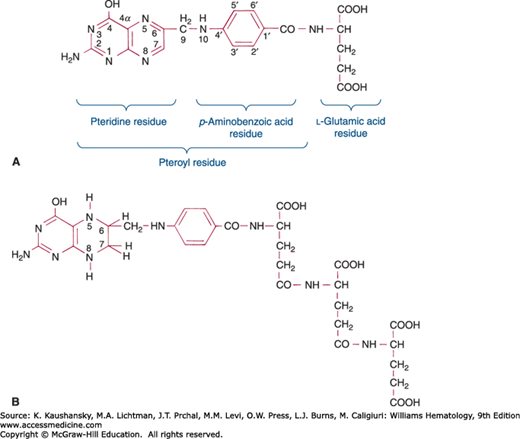
The group of compounds referred to as folates consists of folic acid and its various derivatives. Folic acid (pteroylglutamic acid) is composed of a pteridine moeity, a p-aminobenzoate residue, and an L-glutamic acid residue (Fig. 41–1A). The first two together are called pteroic acid.1 In nature, folates occur largely as conjugates in which multiple glutamic acids are linked by peptide bonds involving their γ-carboxyl groups (Fig. 41–1B). Additionally, the naturally occurring polyglutamated folates are reduced in the 5, 6, 7, and 8 positions of the pteridine ring (as described below, Fig. 41–1B). Conjugates are named according to the length of the glutamate chain (e.g., pteroylmonoglutamate, pteroyldiglutamate, pteroylhexaglutamate). Therapeutic folic acid (pteroylglutamic acid, abbreviated PteGlu, or F) has one glutamic acid and the pteridine ring is not reduced.
To form a functional compound, folate must be reduced to tetrahydrofolate (FH4; see Fig. 41–1B). In this reduction, dihydrofolate (FH2) is an intermediate. A single enzyme, FH2 reductase, catalyzes both F→FH2 and FH2→FH4.
The folate family consists largely of FH4 derivatives bearing a one-carbon substituent (symbolized as FH4-C). The varieties of FH4-C differ with regard to the identity of the one-carbon unit and the site of its attachment to FH4. Figure 41–2 shows one-carbon substituents of biochemical significance and their major interconversions.
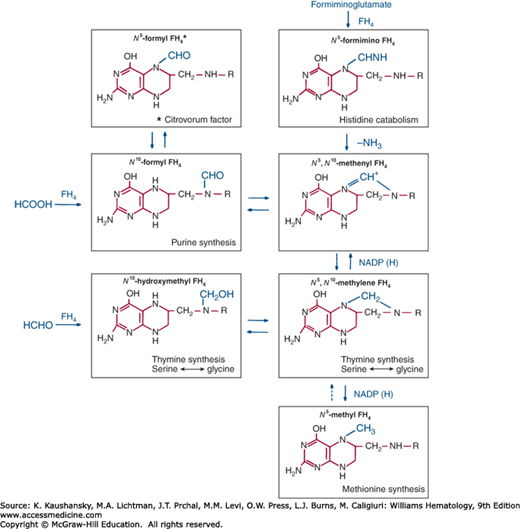
These substituents are attached to FH4 through N5,N10, or both (see Fig. 41–2). Specific enzymes interconvert these various FH4 derivatives through oxidations that require nicotinamide adenine dinucleotide phosphate (NADP) and reductions that utilize the reduced form of NADP, NADPH.2
Reduced derivatives of folic acid usually are sensitive to air oxidation. An important exception is N5-formyl FH4, also called citrovorum factor, leucovorin, or folinic acid, which, because of its stability, is the form preferred for clinical use.
Folic acid comes from many sources. The richest vegetable sources are asparagus, broccoli, endive, spinach, lettuce, and lima beans. Each vegetable contains more than 1 mg of folate per 100 g dry weight. The best fruit sources are oranges, lemons, bananas, strawberries, and melons. Folates also are abundant in liver, kidney, yeast, mushrooms, and peanuts. Since the advent of folic acid fortification of the food supply, the median daily intake of folate from an average American diet is estimated to be 350 mcg.3 Foods are readily depleted of folate by excessive cooking, especially with large amounts of water, discarded before ingestion.
In the normal adult, the minimum daily requirement for folic acid is approximately 50 mcg. The average diet contains many times this amount, but some of the folate may be unavailable. Accordingly, the officially recommended dietary allowance of food folate for an adult is 0.4 mg.3 This figure is derived through considerations of the nutrient requirements to satisfy the needs of 97 to 98 percent of healthy individuals and the relative differences in absorption and bioavailability between dietary folate and the more bioavailable synthetic folic acid. One microgram of food folate is the dietary equivalent of 0.6 mcg folic acid added to food. The body is thought to contain approximately 5 mg of folate.4 When folate intake is reduced to 5 mcg/day, megaloblastic anemia develops in approximately 4 months.5
Folic acid requirements increase in hemolytic anemia, leukemia, and other malignant diseases, in alcoholism6 and during growth; in pregnancy and during lactation requirements increase threefold to sixfold.7 Adequate folate supplies are particularly important in pregnant and lactating women, in whom the recommended daily allowance is increased to 600 and 500 mcg/day, respectively, to meet requirements.8
FH4 is an intermediate in reactions involving the transfer of one-carbon units from a donor to an acceptor. Table 41–1 summarizes the metabolic systems of animal tissues known to require folic acid coenzymes.
| System | Related Transformations of Folic Acid Coenzymes |
|---|---|
| Serine → glycine | Serine + tetrahydrofolate (FH4) → N5,N10-methylene FH4 + glycine |
| Thymidylate synthesis | Deoxyuridylate (dUMP) + N5,N10-methylene FH4 → FH2 + thymidylate (dTMP) |
| Histidine catabolism | Formiminoglutamate + FH4 → N5-formimino FH4 + glutamate |
| Methionine synthesis | Homocysteine + N5-methyltetrahydrofolate → FH4 + methionine |
| Purine synthesis | Glycinamide ribotide + N10-formyl FH4 → FH4 + formylglycinamide ribotide |
| Purine synthesis | 5-Amino-4-imidazole carboxamide ribotide + N10-formyl FH4 → FH4 + 5-formamido-4-imidazolecarboxamide ribotide |
One-carbon units enter the folate pool principally via the serine hydroxymethyltransferase (SHMT) reaction which requires pyridoxal phosphate as a cofactor.9 There are both cytoplasmic (SHMT1) and mitochondrial (SHMT2) isoforms of SHMT which impart a state of functional redundancy for this important enzyme, which is the primary source of one-carbon units for purine and pyrimidine biosynthesis. The cytoplasmic form can undergo sumoylation during S-phase, allowing nuclear localization.10
Among the several one-carbon transfers mediated by folic acid, the transfer that appears to be the most important clinically is the methylation of deoxyuridylate to thymidylate, catalyzed by the enzyme thymidylate synthase.2,10,11 This reaction is an essential step in the synthesis of DNA (Fig. 41–3). In carrying out this reaction, N5,N10-methylene FH4 simultaneously transfers and reduces a one-carbon group, itself serving as the hydrogen donor for the reduction.12 The reaction generates FH2, which must be reduced again to FH4 by FH2 reductase and NADPH before it can again be utilized as a coenzyme:
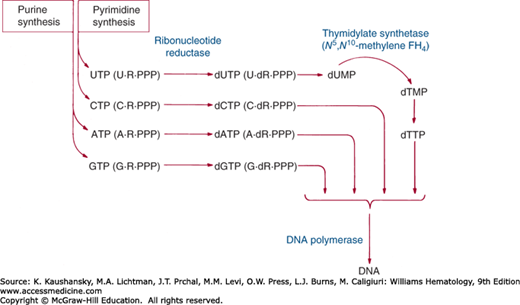
where dUMP = deoxyuridine monophosphate; dTMP = deoxythymidine monophosphate; and NADP = nicotinamide adenine dinucleotide phosphate. Limitation of thymidylate synthesis in folic acid deficiency causes incorporation of uracil instead of thymine into DNA.13
A key enzyme, methylenetetrahydrofolate reductase (MTHFR) regulates the distribution of reduced folates by controlling the rate of NADPH-mediated conversion of N5,N10-methylene FH4 to N-methyltetrahydrofolate. Because N5,N10-methylene FH4 is the obligate one-carbon donor for thymidylate synthesis, its conversion to N-methyltetrahydrofolate serves as a brake on DNA synthesis and repair, while diverting more folate to the methionine synthase reaction (see Fig. 41–2). The activity of MTHFR therefore serves as a checkpoint for intracellular folate trafficking and distribution, and thus becomes more critical in states of folate depletion.
A polymorphic form of MTHFR, MTHFR 677C→T, is of some clinical importance. The mutation results in a thermolabile form of the enzyme with a higher Km (Michaelis-Menten dissociation constant) for its methylene-FH4 substrate. Retardation of the folate methylation cycle makes more methylene-FH4 available for thymidylate synthesis (Fig. 41–4), and also affects the levels of homocysteine, an amino acid whose rate of production depends on both folate and cobalamin.
Folate deficiency diminishes purine biosynthesis by slowing (1) the folate-dependent formylation of glycinamide ribotide to N-formylglycinamide ribotide, the reaction that places the C-8 in the purine ring, and (2) the folate-dependent conversion of 5-amino-4-imidazole carboxamide ribotide (AICAR) to 5-formamido-4-imidazole carboxy-amide ribotide, the reaction that places the C-2 in the purine ring.14 Additional reactions dependent on biopterin, a nonfolate pteridine derivative, that are of potential metabolic importance are hydroxylation of phenylalanine to tyrosine, oxidation of long-chain alkyl ethers of glycerol to fatty acid, hydroxylation of tryptophan to 6-hydroxytryptophan (a precursor of serotonin), 17α-hydroxylation of progesterone,15 and production of nitric oxide.16 Tetrahydrofolic acid is weakly active in some of these systems in vitro17; whether it plays any such role in vivo is unknown.
Intracellular folates exist primarily as polyglutamate conjugates.18 Approximately 75 percent of the folate in human erythrocytes and leukocytes is polyglutamylated.19 Plasma folate consists largely of the monoglutamate N5-methyltetrahydrofolate and is transported into the cells in this form.20 Inside the cells, the polyglutamate chain is sequentially built up by an ATP-dependent folylpoly-γ-glutamyl synthase (FPGS).21 The activity of human FPGS depends strongly on the form of the folate substrate, declining in the order FH4 > N10-formyl FH4 > N5-methyltetrahydrofolate, toward which the enzyme is almost inert.22 In humans, conjugated folates carry on average seven to eight glutamyl residues.23 Intracellular folylmonoglutamates leak out of the cells at a fairly rapid rate whereas polyglutamates do not, presumably because of the highly charged polyglutamate tail.24 Therefore, attachment of the polyglutamate chain is essential for retaining folates within cells. Folylpolyglutamates are superior to monoglutamates as substrates for folate-dependent enzyme reactions.19
The duodenum and proximal jejunum is the principal site of folate absorption. Absorption of a dose of either monoglutamate or polyglutamate forms of folate begins within minutes. Peak plasma levels are reached in 1 to 2 hours. Because only folylmonoglutamate appears in plasma, all folylpolyglutamates must first be hydrolyzed by the enzyme glutamate carboxypeptidase II (folate hydrolase)25 during absorption across the intestine.26 This enzyme (previously referred to as “conjugase”) is located on the brush-border membrane and plays an important role in the intestinal absorption of folate.27 Folylpolyglutamate is hydrolyzed at the brush-border of the intestinal cell (Fig. 41–5). Folate hydrolase purified from human jejunum catalyzes the Zn2+-dependent deconjugation of folate polyglutamates ranging from PteGlu2 to at least PteGlu7.28 It is an exopeptidase that successively removes single glutamate residues from the end of the polyglutamate chain, ultimately yielding the folylmonoglutamate. The monoglutamate forms are then taken up by one of two folate-specific transporters located on the apical brush-border, the reduced folate carrier (RFC) or the proton-coupled folate transporter (PCFT).25 Although RFC has a pH optimum of 7.4, PCFT is a high-affinity folate transporter that uses a proton-coupled system to facilitate folate absorption and shows maximum transport activity at a low pH.25,29 This property is consistent with the observation that cancer cells retain a high affinity for the new-generation antifolate pemetrexed. Defects in the PCFT are the underlying cause of hereditary folate malabsorption.30
Figure 41–5.
Digestion and absorption of folate polyglutamate by the intestine. The polyglutamate (in this case, PteGlu7) is hydrolyzed in the intestinal lumen or at the brush-border. The resulting pteroylglutamate (PteGlu) is transported into the intestinal cell, where it is reduced and methylated, appearing in the circulation chiefly as N5-methyltetrahydrofolate.
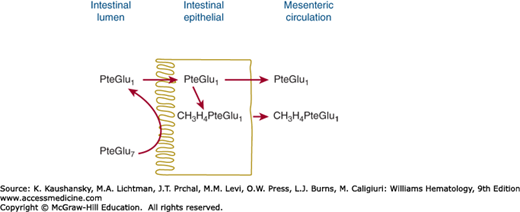
Folate hydrolases also are found outside the intestine. For example, human plasma contains sufficient hydrolase activity to convert polyglutamates containing more than three glutamyl residues to monoglutamates. Other γ-glutamyl hydrolases appear to be lysosomal carboxypeptidases31 that are not involved in absorption of folates from the intestine but that play a role in the release of folate from storage sites in the liver and kidney.25
Folate monoglutamates are actively transported across the intestinal epithelium by PCFT-mediated transport (Km = 1 to 2 μM) that is independent of Na+, K+, and transmembrane potential.32 The mechanism uses the pH gradient between the jejunal lumen (pH ~6) and the interior of the epithelial cell to drive folate into the cell against a concentration gradient.33 Passive transport also may occur.34 In the intestinal cell, the absorbed folate monoglutamates are reduced if necessary, and then converted to N5-methyltetrahydrofolate (some N10-formyl FH4 also is made) and transported into the bloodstream without further change.35
Folate undergoes an enterohepatic cycle in which it is first secreted against a concentration gradient into the bile, appearing there chiefly as N5-methyltetrahydrofolate monoglutamate, and then is reabsorbed from the small intestine.36 Bile contains approximately two to 10 times the folate concentration of normal serum, with biliary excretion accounting for up to 0.1 mg of folate per day. This quantity is sufficiently large that interruption of the enterohepatic cycle by biliary diversion causes serum folate levels to fall by more than 50 percent in less than 1 day.37 The enterohepatic cycle has been proposed to redistribute folate between hepatic stores and peripheral tissues according to the state of the exogenous folate supplies.38
Tritiated folylmonoglutamate (3H-F) administered intravenously is almost completely removed from the bloodstream in a few minutes.39 Uptake involves two classes of folate-binding proteins40: high-affinity folate receptors41 that concentrate folate in intracellular vesicles and a membrane folate transporter that transports folate from the vesicles into the cytosol. The high-affinity receptors, which are attached to the outer surface of the cell membrane by glycosyl-phosphatidylinositol linkages and lack an intracytoplasmic portion,42 bind very tightly (Kd [distribution coefficient] in the nanomolar range) to most physiologic folate monoglutamates,43 particularly N5-methyltetrahydrofolate, the major circulating folate.44 The folate receptors exist in various isoforms (of which α and β are the most important). Folate receptor-α, despite its missing cytoplasmic extension, is effective in mediating endocytosis.45 Their very high affinity enables the receptors to take up N5-methyltetrahydrofolate from the plasma, even at its ambient concentration of approximately 10 nM. The membrane folate transporter is a probenecid-inhibitable organic anion carrier that, among other functions, carries reduced folates and methotrexate in and out of the cytoplasm.40 Its Km for folate is in the micromolar range. Once internalized, the folates are retained by the cells partly through polyglutamylation46 as well as through tight association with a set of intracellular folate-binding proteins.47 Three of these proteins are enzymes involved in methyl group metabolism: sarcosine dehydrogenase and dimethylglycine dehydrogenase (mitochondrial)48 and glycine N-methyl transferase (cytosolic).49 Why these enzymes bind folate so avidly or whether this binding affects overall methyl group metabolism is unknown, although glycine N-methyl transferase is speculated to regulate methyl group metabolism by controlling the tissue concentration of S-adenosylhomocysteine (SAH), one of its reaction products and a potent inhibitor of most methyltransferases.
Folates have been found in all body tissues that have been analyzed. The principal form of the vitamin in tissues and in blood appears to be the N5-methyl form.50 The total folate pool turns over very slowly.51 Degradation accounts for a portion of this turnover. p-Aminobenzoylglutamate has been identified as a breakdown product. The fate of the pteridine moiety is unknown.
The soluble folate-binding proteins of serum and milk are high-affinity folate receptors that are released from cell membranes by proteolysis.52 These proteins can be detected in approximately 15 percent of normal individuals53 and are found at increased levels in some pregnant women, women taking oral contraceptives, folate-deficient alcoholics (but not patients with cobalamin deficiency),54 and patients with uremia, hepatic cirrhosis, and chronic myelogenous leukemia.55 In normal subjects, the proteins are approximately two-thirds saturated and have a total folate-binding capacity of approximately 175 pg/mL of serum.56 The proteins may not be detected in some subjects because of complete saturation of the proteins with endogenous unlabeled folate.57 Serum folate-binding protein has an Mr of 40,000 and prefers oxidized to reduced folates.55
Folate-binding proteins have been found in milk and in normal granulocytes.58 Folate bound to the milk folate binder in suckling animals is absorbed chiefly in the ileum59 rather than the jejunum, the principal site of absorption of free folate. The milk folate binder, a glycoprotein, also promotes folate transport into the liver via the asialoglycoprotein receptor.60 The milk folate binder is speculated to protect an infant’s folate supply by preventing bacteria from sequestering the vitamin away from the intestinal absorptive surface. The folate-binding protein in granulocytes has been localized to the specific granules, from which it is released when the granulocytes are stimulated.
Folates are both resorbed and secreted by the kidney. Resorption is accomplished by a membrane-bound high-affinity folate receptor (Km for N5-methyltetrahydrofolate = 0.4 nM) located in the brush-borders of the proximal tubules.61 Filtered folate may thus be returned to the bloodstream. There is resorption of most, but not all, of the filtered folate.
In humans, intact folates and their cleavage products are excreted by the kidney at a rate of 2 to 5 mcg/day.62 A small percentage of parenterally administered labeled folate is recoverable in the feces and mainly represents unreabsorbed folate from the enterohepatic cycle.63
Folates are measured by chemiluminescent methods using various folate binders. These assays are identical in principle to the radioligand binding assays that they have replaced.
COBALAMIN
The cobalamin molecule has two major portions: a porphyrin-like near-planar macrocycle known as corrin, and a nucleotide that lies almost perpendicular to the corrin ring (Fig. 41–6). The corrin moiety contains four reduced pyrrole rings that bind a central cobalt atom whose two remaining coordination positions are occupied by a 5,6-dimethylbenzimidazolyl (5,6-DMB) group, below the ring and various ligands (in this case, —N in the form of CN) above the ring.64
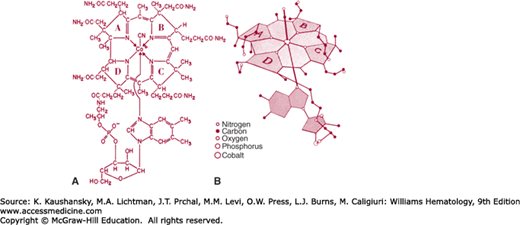
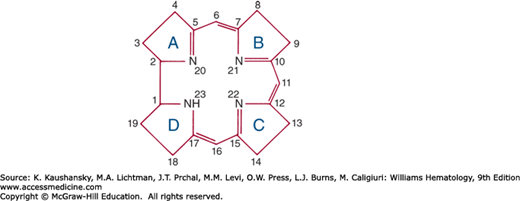
Compounds containing the corrin ring are known as corrinoids. The cobalamins are corrinoids whose nucleotide contains 5,6-DMB. Two connections exist between the corrin and the nucleotide: (1) a bond between the nucleotide phosphate and a side chain in ring D, and (2) a bond between cobalt and a nitrogen atom of benzimidazole. Figure 41–7 summarizes the numbering and ring designations of the corrin system.
The term vitamin B12 is sometimes used as a generic term for the cobalamins. The term probably is best reserved, however, as an alternative name for cyanocobalamin, the usual therapeutic form of cobalamin.
Four cobalamins are important in animal cell metabolism. Two are cyanocobalamin (CnCbl; vitamin B12) and hydroxocobalamin (OHCbl) or aquocobalamin (HOH Cbl). The other two cobalamins are alkyl derivatives that are synthesized from OHCbl and serve as coenzymes. In one, adenosylcobalamin (AdoCbl), a 5′-deoxyadenosyl replaces OH as the cobalt ligand above the ring (Fig. 41–8).65 In the second, methylcobalamin (MeCbl), the upper ligand is a methyl group. MeCbl is the major form of cobalamin in human blood plasma.66
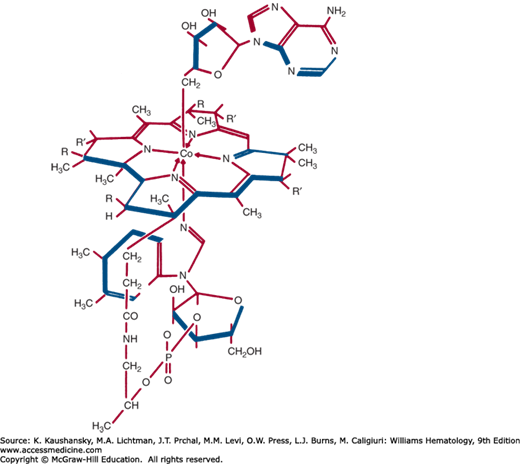
Cobalamin is synthesized only by certain microorganisms; animals ultimately depend on microbial synthesis for their cobalamin supply. Foods that contain cobalamin are of animal origin: meat, liver, seafood, and dairy products. Cobalamin has not been found in plants.
The average daily diet in Western countries contains 5 to 30 mcg of cobalamin, of which 1 to 5 mcg is absorbed.67 Less than 250 ng appears in the urine; the unabsorbed remainder appears in the feces. Total body content is 2 to 5 mg in an adult,68 with approximately 1 mg in the liver. The kidneys also are rich in cobalamin.69 Relative to the daily requirement, body reserves of cobalamin are much larger than those of folate.
Cobalamin has a daily rate of obligatory loss of approximately 0.1 percent of the total-body pool, irrespective of the pool size. For this reason, a deficiency state does not develop for several years after cessation of cobalamin intake. The officially recommended dietary allowance (RDA) for adults is 2.4 mcg2; growth, hypermetabolic states, and pregnancy increase daily requirements. The RDA for children ages 1 to 13 years is 0.9 to 1.8 mcg. Because of insufficient data, no RDA has been established for infants. Instead, adequate intakes of 0.4 mcg for age 0 to 6 months and 0.5 mcg for age 7 to 12 months have been estimated.
The only two recognized cobalamin-dependent enzymes in human cells are AdoCbl-dependent methylmalonyl CoA (coenzyme A) mutase and MeCbl-dependent methyltetrahydrofolate-homocysteine methyltransferase.
Methylmalonyl CoA mutase is a mitochondrial enzyme that participates in the disposal of the propionate formed during breakdown of valine, isoleucine, and odd-carbon fatty acids. The enzyme is a homodimer of a 78-kDa subunit that is encoded by a gene on chromosome 6.70 In the reaction catalyzed by methylmalonyl CoA mutase, methylmalonyl CoA, which is produced during catabolism of propionate,71 is converted to succinyl CoA, a Krebs cycle intermediate. In the course of this reaction, a hydrogen on the methyl carbon of the substrate exchanges places with the —COSCoA group (Fig. 41–9).
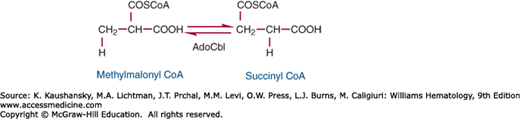
The coenzyme serves as an intermediate hydrogen carrier, accepting the hydrogen from the substrate in the initial phase of the reaction and returning it to the product after migration of —COSCoA.
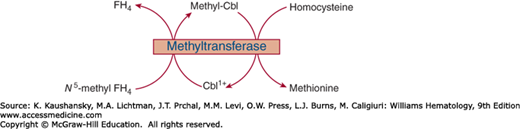
MeCbl participates in cobalamin-dependent synthesis of methionine from homocysteine by the enzyme N5-methyltetrahydrofolate–homocysteine methyltransferase. S-adenosylmethionine (SAMe) and a second enzyme, methionine synthase reductase are required for methyltransferase activity.72 The reductase converts the oxidized cobalt to the readily alkalizable Co+, which then accepts a methyl group from SAMe, a powerful biologic methylating agent, thereby restoring activity of the methyltransferase. In humans, this pathway also serves as a mechanism critical for converting N5-methyltetrahydrofolate to FH4 required for synthesis of polyglutamates as well as other important one-carbon adducts of folate. The demethylation of N5-methyltetrahydrofolate is a prerequisite for attachment of the polyglutamate chain to newly acquired folate, which is largely taken up by the cell in the form of N5-methyltetrahydrofolate monoglutamate.24 Nitrous oxide (N2O) impairs the methyltransferase by oxidizing cob(I)alamin (a catalytic intermediate in the methyltransferase reaction) to cob(II)alamin. This reaction depletes MeCbl and produces a cobalamin deficiency-like state.
Because cobalamin has the capacity to bind cyanide, it may participate in detoxification of cyanide. Tobacco and certain foods (fruits, beans, tubers, and nuts) contain cyanide in the form of thiocyanate. Although the evidence is inconclusive, cobalamin is believed to play a role in neutralizing cyanide taken in via these substances.64
In both folate deficiency and cobalamin deficiency, the megaloblastic anemias are fully corrected by treatment with the appropriate vitamin. The megaloblastic anemia of cobalamin deficiency also is variably corrected by folic acid supplementation even if no cobalamin is given, although the remission may be partial and only temporary. Conversely, the anemia of folate deficiency is generally not helped at all by cobalamin although partial responses to high doses of cobalamin have been reported in some patients with folate deficiency.73 These clinical observations indicate that the megaloblastic anemia in cobalamin deficiency actually results from an abnormality in folate metabolism.24 The observation that urinary excretion of formiminoglutamic acid (FIGlu) and AICAR, normally regarded as a sign of folate deficiency, is seen occasionally in pure cobalamin deficiency74 provides further evidence that folate metabolism is deranged by cobalamin deficiency. Two explanations have been proposed to account for the folate responsiveness of cobalamin-deficient megaloblastic anemia: (1) the methylfolate trap hypothesis, which is accepted by the majority of authorities, and (2) the formate starvation hypothesis (Fig. 41–10).
Figure 41–10.
Methods by which cobalamin deficiency decreases intracellular folate levels. Methyltetrahydrofolate (MeFH4), the principal form of folate in the bloodstream, circulates in the unconjugated form (i.e., it has no polyglutamate side chain). This and other forms of unconjugated FH4 can be taken into cells but leak out again unless they are conjugated. MeFH4 is not a substrate for the conjugating enzyme, so conjugation cannot occur until the MeFH4 is converted to another form of folate. Cobalamin is necessary for this process because it is the cofactor for the reaction that converts MeFH4 to FH4. In cobalamin deficiency, the conversion of MeFH4 to FH4 is defective. Newly transported folate remains in the form of MeFH4, which cannot be conjugated and leaks back out of the cell. A. According to the methylfolate trap hypothesis, all forms of FH4 other than MeFH4 can be conjugated, so MeFH4 is the only folate species that leaks out of the cell. B. The formate starvation hypothesis differs from the methylfolate trap hypothesis solely in assuming that only the formylated folates (N10-formyl FH4 and/or N5,N10-methenyl FH4) can be conjugated, so newly transported MeFH4, N5,N10-methylene FH4 and free FH4 leak out of the cell. (CHO) FH4 = N10-formyl FH4. Homocys met, homocysteine methyltransferase.
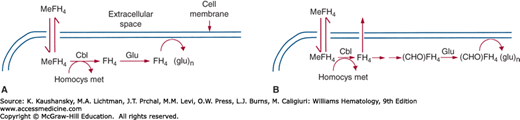
The methylfolate trap hypothesis75 is based on the fact that the folate-requiring enzyme N5-methyltetrahydrofolate–homocysteine methyltransferase is also dependent on cobalamin. The hypothesis states that in cobalamin deficiency tissue folates are gradually diverted into the N5-methyltetrahydrofolate pool because of slowing of the methyltransferase reaction,76 the only route out of that pool for folate. As N5-methyltetrahydrofolate levels increase, the levels of other forms of folate decline, with a consequent fall in the rates of reactions in which those forms participate. In particular, because the MTHFR reaction is irreversible, methylene-FH4 becomes depleted, the synthesis of dTMP is slowed, and megaloblastic anemia ensues.
In its simplest form, the hypothesis predicts that in cobalamin deficiency tissue levels of N5-methyltetrahydrofolate are abnormally high and those of other forms of folate are abnormally low. Although serum N5-methyltetrahydrofolate levels are frequently elevated in cobalamin deficiency,77 tissue folate levels, predominantly polyglutamates, decline.78 The decreased level appears to be related to the substrate specificity of the folate-conjugating enzyme. This enzyme works very poorly with N5-methyltetrahydrofolate; therefore, it is unable to carry out normal γ-glutamylation of newly internalized N5-methyltetrahydrofolate monoglutamate in cobalamin-deficient cells because the freshly acquired folate cannot be converted into a suitable substrate (i.e., free FH4 or formyl FH4). Thus, although sequestration of tissue folates in an expanded N5-methyltetrahydrofolate pool may account for some of the effects of the blockade in methyltransferase activity, the major problem seems to be a failure to convert newly acquired folate into a form that can be retained by the cell. The upshot is development of tissue folate deficiency as the unconjugated folate leaks out (see Fig. 41–10). The whole process is aggravated by a drop in tissue levels of SAMe as the methionine supply is curtailed because of the diminished activity of the methyltransferase.79 SAMe, which is necessary for methyltransferase activity, is also a powerful inhibitor of N5,N10-methylene FH4 reductase MTHFR,80 the enzyme responsible for production of N5-methyltetrahydrofolate. The relief of this inhibition as SAMe levels fall accelerates the flow of folates toward N5-methyltetrahydrofolate, further aggravating the metabolic imbalance resulting from impairment in methyltransferase activity.
This problem could be overcome if N5-methyltetrahydrofolate were converted into a substrate for the conjugating enzyme by another route. In theory, this could be accomplished by reversal of the N5,N10-methylene FH4 reductase reaction. For practical purposes, however, the N5,N10-methylene FH4 reductase reaction is irreversible in vivo.81
This hypothesis holds that formate starvation is the basis for folate-responsive megaloblastic anemia of cobalamin deficiency.82 This theory is based on the diminished capacity of cobalamin-deficient lymphoblasts to incorporate formaldehyde into purine and methionine79 and on experiments showing that N5-formyl FH4 is more effective than FH4 at correcting some of the abnormalities in folate metabolism seen in cobalamin deficiency.83 The hypothesis states that with the decrease in methionine production in cobalamin-deficient conditions, the generation of formate is depressed (because normally the methyl group of excess methionine is rapidly oxidized to formate),84 leading to a decline in the production of N5-formyl FH4.
Intrinsic factor is one of a number of binding proteins in which cobalamin is ensconced as it makes its way through the body (Table 41–2). Intrinsic factor is needed for the absorption of cobalamins taken orally at physiologic dosage levels. Human intrinsic factor is a glycoprotein (Mr approximately 44,000) encoded by a gene on chromosome 11.85 It has binding sites for cobalamin and a specific ileal receptor, the former situated near the carboxy-terminus and the latter near the aminoterminus of the intrinsic factor molecule.86 Binding to cobalamin is very tight, and involves the 5,6-DMB lower axial ligand of the molecule. This specificity allows for the exclusion of other noncobalamin corrinoids during the tightly regulated absorptive process.64 The entrapment of the vitamin alters the conformation of intrinsic factor, producing a more compact form that is resistant to proteolytic digestion.
| Protein | Source | Function |
|---|---|---|
| Intrinsic factor | Gastric parietal cells | Promotes absorption uptake of cobalamin by ileum |
| Transcobalamin | Probably all cells | Promotes uptake of cobalamin by cells |
| Haptocorrin | Exocrine glands, phagocytes | Helps dispose of cobalamin analogues (?) |
In humans, intrinsic factor is synthesized and secreted by the parietal cells of the cardiac and fundic mucosa.87 Secretion of intrinsic factor usually parallels that of hydrochloric acid (HCl). It is enhanced by the presence of food in the stomach, vagal stimulation, histamine, and gastrin. Gastric juice also contains other cobalamin-binding glycoproteins.88 These proteins were known as the R proteins because of their rapid electrophoretic mobility compared with intrinsic factor. Elucidation of the primary protein structure of the R proteins reveals that they belong to the same family of isoproteins as the plasma haptocorrin (HC) binder (previously known as transcobalamins I and III). These HC-like proteins are produced mainly by the salivary glands.
Cobalamins in foods are liberated in the stomach by peptic digestion.89 They are then bound not to intrinsic factor but to the HC-like protein because cobalamin binds much more tightly to HC than to intrinsic factor at the acid pH of the stomach.90 Upon entering the duodenum, cobalamin is released from the cobalamin–HC protein complex through digestion by pancreatic proteases, which in normal subjects act by selectively degrading HC and the cobalamin–HC complex while sparing intrinsic factor.90 Only at this point can cobalamin bind to intrinsic factor to form the intrinsic factor–cobalamin complex.
The intrinsic factor–cobalamin complex, which is very resistant to digestion,91 traverses the intestine until it reaches the intrinsic factor receptor, cubilin,92 a 460-kDa peripheral membrane glycoprotein located in the microvillus pits of the ileal mucosa brush-border. Cubilin forms part of a multifunctional epithelial receptor complex also found in the yolk sac and renal proximal tubule cells.93 In the kidney, it appears to serve a role in the overall body economy through tubular reabsorption of cobalamin,94 but the function of the cubilin receptor complex in the kidney and other polarized epithelial surfaces extends beyond cobalamin. The ileal cubilin receptor complex consists of two proteins, cubilin (CUB) and amnionless (AMN), the product of two distinct genes, CUB and AMN. Both proteins, which together have been designated the “CUBAM complex,” colocalize in the endocytic compartment and are required for the process of assimilation of cobalamin,95 AMN serving as a chaperon for endosomal targeting. Mutations affecting either of the two proteins disrupt the normal process of the intestinal phase of cobalamin absorption. In addition to the tightly embracing components of the CUBAM complex, a distinct large multifunctional protein, megalin, which belongs to the low-density lipoprotein family,96 also participates in the conformational changes that accompany internalization. The concentration of the CUBAM complex rises progressively to a maximum near the terminal ileum.97 A specific site on the intrinsic factor molecule avidly attaches to the receptor in a binding reaction that requires a pH of 5.4 or greater and Ca2+ (or other divalent cations) but no energy.98
The intrinsic factor–cobalamin receptor complex is taken into the ileal mucosal cells over 30 to 60 minutes by endocytosis,99 where the vitamin is processed and released into the portal blood over many hours. The receptors recycle to the microvillus surface to shuttle another load of intrinsic factor–cobalamin complex.99 That this process has a limited capacity is evident from estimates of the maximum amount of cobalamin that can be absorbed from a single dose via this physiologic pathway.64,100 Defects in the genes that regulate the complex mechanism of ileal absorption are implicated in autosomal recessive megaloblastic anemia (MGA1), caused by intestinal malabsorption of cobalamin (see “Selective Malabsorption of Cobalamin, Autosomal Recessive Megaloblastic Anemia, Imerslund-Gräsbeck Disease” below).
During its sojourn in the ileal enterocyte, the vitamin first appears in the lysosomes, but by 4 hours most of the vitamin is located in the cytosol.101 During absorption, the entire intrinsic factor–cobalamin complex appears to be taken into the cell, where the cobalamin is released while the intrinsic factor is degraded.102
Cobalamin from a small oral dose (10 to 20 mcg) starts to appear in the blood after 3 to 4 hours, and the vitamin reaches a peak level in 6 to 12 hours. In the portal blood, the cobalamin is complexed with a cobalamin-transporting protein known as transcobalamin (TC) previously known as TC II.103 There is evidence that cobalamin leaves the enterocyte through a portal that is part of the ABC drug transport system, ABCC1 (also known as the multidrug resistance protein1 [MRP1]) located on the basolateral surface of the intestinal epithelium, as well as other polar cells and nonpolar cells (macrophages).104 The cobalamin–TC complex is now believed to be formed as it exits the ileal enterocyte, one of a variety of cells that synthesize TC, including the neighboring vascular endothelial cells in the submucosa.105 Large oral doses (1 mg) of cobalamin are absorbed inefficiently (1 to 2 percent of an oral dose) by simple diffusion that is not mediated by intrinsic factor.100 In these instances, the vitamin appears in blood within minutes, again as the cobalamin–TC complex.
Like the folates, the cobalamins undergo appreciable enterohepatic recycling.106 In humans, between 0.5 and 9 mcg/day of cobalamin is secreted into the bile, where it is bound to HC.107 After entering the intestine, the cobalamin–HC complexes of biliary origin are treated exactly like those delivered from the stomach. The cobalamin is released by digestion of the HC by pancreatic proteases, and then is taken up by intrinsic factor and reabsorbed. From 65 to 75 percent of biliary cobalamin is estimated to be reabsorbed by this mechanism.108 Because of the size of the cobalamin storage pool and the existence of this enterohepatic circulation, a very long time—as long as 20 years—is required for a clinically significant cobalamin deficiency to develop from a diet providing insufficient cobalamin (e.g., a strictly vegetarian diet).109 Patients who are unable to absorb the vitamin, however, become clinically deficient in only 3 to 6 years because the absorption of both biliary and dietary cobalamin are interdicted.110
TC is the plasma protein that mediates the transport of cobalamin into the tissues.111 A β-globulin protein with a calculated molecular weight of 45,538 from the deduced amino acid sequence,112,113 TC binds cobalamin with exceedingly high affinity (Ka = 10–11 M).114 Unlike intrinsic factor, whose binding is highly specific for cobalamins, TC shows some promiscuity and also can bind certain corrins that are chemically related to the cobalamins but have no function in mammalian systems and are known as cobalamin “analogues.”115 TC is synthesized by many types of cells, including enterocytes, hepatocytes, endothelial cells, mononuclear phagocytes, fibroblasts, and hematopoietic precursors in the marrow.64 Although circulating TC carries only a minor fraction of the cobalamin in the plasma, it is the protein on which cobalamin absorbed through the intestine and is transported into the portal blood as the preformed cobalamin–TC complex. It also is the protein with which cobalamin given parenterally associates almost immediately.116 These cobalamin–TC complexes are transported into the tissues within minutes of appearing in the bloodstream.117 The transport process begins with binding of the cobalamin–TC complex to a specific membrane receptor that is present on a wide variety of cells.118 The protein and gene encoding the TC receptor has been purified from placental membranes and characterized.119 Designated as CD320, the receptor belongs to the low-density lipoprotein receptor family and its internalization involves megalin. The receptor-bound complex is internalized by receptor-mediated endocytosis and delivered to a lysosome, where the TC is digested and the cobalamin is freed.120,121
To become metabolically active, CnCbl and OHCbl must first be converted to AdoCbl and MeCbl, the coenzymatically active cobalamins. The conversion is accomplished by reduction and alkylation. CnCbl and OHCbl are first reduced to the Co++ form [cob(II)alamin] by NADPH- and NADH (nicotinamide adenine dinucleotide phosphate)-dependent reductases that are present in mitochondria and microsomes.122 CN– and OH– are displaced from the metal during reduction. Some of the cob(II)alamin is reduced further in the mitochondria to the intensely nucleophilic Co+ form [cob(I)alamin]. This is then alkylated by ATP to form AdoCbl in a reaction in which the 5′-deoxyadenosyl moiety of ATP is transferred to the cobalamin and the three phosphates of ATP are released as inorganic triphosphate (Fig. 41–11). The rest of the cobalamin binds to cytosolic N5-methyltetrahydrofolate–homocysteine methyltransferase, where it is converted to MeCbl. The several steps involved in the conversion of cobalamin to its coenzymatically active forms are regulated by genes that play a critical role in the processing of the vitamin. There are a number of inherited metabolic errors that correspond to one or more of these specific steps and that result in characteristic syndromes affecting aspects of cobalamin metabolism that are discussed later in this chapter.
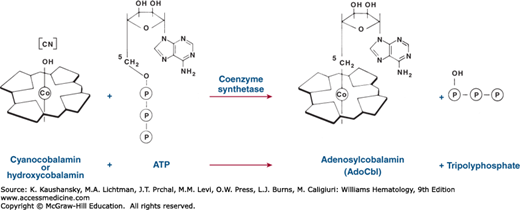
The HCs (previously known as R proteins) are a group of immunologically related proteins of apparent Mr approximately 60,000, consisting of a single polypeptide species variably substituted with oligosaccharides that terminate with different quantities of sialic acid.123 They are found in milk, plasma, saliva, gastric juice, and numerous other body fluids. They appear to be synthesized by mucosal cells of the organs that secrete them124 and by phagocytes.125 Although the HCs bind cobalamin, they lack intrinsic factor activity, that is, they are unable to promote the intestinal absorption of the vitamin.
Plasma HC carries most (70 to 90 percent) of the circulating cobalamin. It contains nine potential glycosylation sites126 and is encoded by a gene on chromosome 11, the same chromosome that carries the intrinsic factor gene.127 In contrast to TC, HC clearance from the plasma is very slow (half-life [T1/2]: 9 to 10 days).128 The asialoglycoprotein receptor carries the cobalamin–HC complexes into the hepatocytes, where they are chiefly eliminated. The complexes are degraded, and their load of cobalamins is excreted in the bile.106,129 HC binds its ligands more tightly than does either intrinsic factor or TC. Furthermore, HC is less restrictive than either intrinsic factor or TC with respect to ligand specificity; it avidly takes up corrinoids of widely varying structure.130 The ligand-binding properties of HC and its mode of clearance by the liver suggest that HC helps clear the system of nonphysiologic cobalamin analogues that may have been acquired or may have arisen through degradation of cobalamin.131,132 As the liver metabolizes analogue–HC complexes, it secretes the analogues into the bile. Because these analogues are bound poorly by intrinsic factor,130 they are poorly reabsorbed from the intestine and are eliminated in the feces. The precise role of HC is unknown, although it may play a role in the body economy of cobalamin by facilitating excretion of cobalamin analogues while conserving cobalamin through enterohepatic recycling. Additionally, it has been proposed that HC may serve an antimicrobial role.64
As with folate, cobalamin is usually measured with automated competitive displacement assays using intrinsic factor as a cobalamin-binding protein. The misleading results previously provided by competitive ligand displacement assays were explained by the discovery in serum and tissue of a class of cobalamin analogues that are detected by the radioisotope assay when HC-type proteins and not intrinsic factor were used as the binding protein.133 Current assays use intrinsic factor as the binder and give more reliable values for serum cobalamin. The chemical nature and biologic significance of the analogues are unknown,134 but recent evidence suggests that they may arise in the gastrointestinal tract.131,132
TC and HC are present in plasma in trace quantities (approximately 7 and 20 mcg/L, respectively). In fasting plasma, at least 70 percent of the circulating cobalamin is bound to HC.135 TC binds only 10 to 25 percent of the total plasma cobalamin,136 but provides the majority (approximately 75 percent) of the total unsaturated cobalamin-binding capacity of plasma.135 Table 41–3 lists alterations in unsaturated cobalamin-binding capacity and in HC and TC levels in various disease states. In recent years, assays have been developed that measure the fraction of the plasma cobalamin that is bound to TC. This component, known as holotranscobalamin (holoTC), shows improved specificity compared with the standard cobalamin assay for identifying true cobalamin deficiency, although the assays appear to be generally comparable with respect to sensitivity.137,138,139,140,141,142,143
| Binder | Disease |
|---|---|
| Increased HC (TCI, R protein) | Myeloproliferative disorders |
| Polycythemia vera | |
| Myelofibrosis | |
| Benign neutrophilia | |
| Chronic myelocytic leukemia | |
| Hepatoma (occasionally) | |
| Metastatic cancer | |
| Increased TC | Myeloproliferative disorders |
| Liver disease | |
| Inflammatory disorders | |
| Gaucher disease | |
| Anti-TC antibodies | |
| Unsaturated cobalamin binders | |
| Increased | Transient neutropenia |
| Elevated HC | |
| Decreased | Liver disease |
| Elevated serum cobalamin |
MEGALOBLASTIC ANEMIAS
Megaloblastic anemias are disorders caused by impaired DNA synthesis. The presence of megaloblastic cells is the morphologic hallmark of this group of anemias. Megaloblastic red cell precursors are larger than normal and have more cytoplasm relative to the size of the nucleus. Promegaloblasts show a blue granule-free cytoplasm and a fine “salt and pepper” granular chromatin that contrasts with the ground-glass texture of its normal counterpart. As the cell differentiates, the chromatin condenses more slowly than normal into darker aggregates that coalesce, but do not fuse homogeneously, giving the nucleus a characteristic fenestrated appearance. Continuing maturation of the cytoplasm as it acquires hemoglobin contrasts with the immature-looking nucleus—a feature termed nuclear-cytoplasmic asynchrony.
Megaloblastic granulocyte precursors are also larger than normal and show nuclear-cytoplasmic asynchrony. A characteristic cell is the giant metamyelocyte, which has a large horseshoe-shaped nucleus, sometimes irregularly shaped, containing ragged open chromatin.
Megaloblastic megakaryocytes may be abnormally large and polylobated, with deficient granulation of the cytoplasm. In severe megaloblastosis, the nucleus may show detached lobes. Further details are provided in “Laboratory Features” below and in Figs. 41–12 and 41–13.
Figure 41–12.
A. Pernicious anemia. Blood film. Note the striking oval macrocytes, wide variation in red cell size, and poikilocytes. Despite the anisocytosis and microcytes, the mean red cell volume is usually elevated, as in this case (mean corpuscular volume [MCV] = 121 fL). B. Marrow precursors in pernicious anemia. Note very large size of erythroblasts (megaloblasts) and asynchronous maturation. Cell on right is a polychromatophilic megaloblast with an immature nucleus for that stage of maturation. Cell on left is an orthochromatic megaloblast with a lobulated immature nucleus. An orthochromatic megaloblast with a condensed nucleus is between and above those two cells. C and D. Two examples of hypersegmented neutrophils characteristic of megaloblastic anemia. The morphology of blood and marrow cells in folate-deficient and vitamin B12-deficient patients is identical. The extent of the morphologic changes in each case is related to the severity of the vitamin deficiency. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
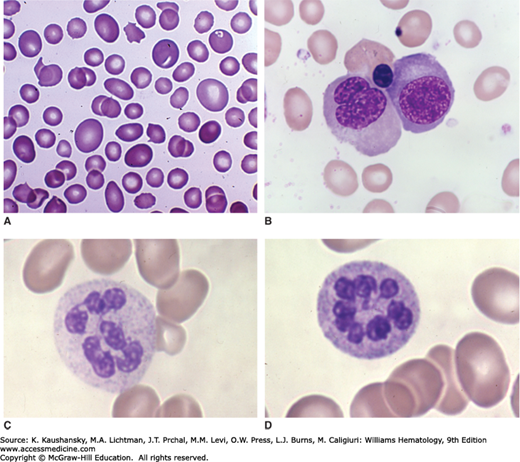
Figure 41–13.
Marrow films. Megaloblastic anemia. Patient with pernicious anemia (vitamin B12 deficiency). A. Basophilic megaloblasts. Large cell size, very characteristic nuclear chromatin pattern with exaggerated proportion of euchromatin. B. Polychromatophilic megaloblast. Very large cell size for maturational stage. Large nuclear size and abnormally large proportion of euchromatin without appropriate nuclear condensation at this stage of maturation. Adjacent lymphocyte. C. Polychromatophilic megaloblast with small nuclear fragment. Arrow indicates giant band neutrophil. At lower left is orthochromatic megaloblast with multiple nuclear fragments. D. Oblique arrow indicates promegaloblast. Horizontal arrow indicates giant band neutrophil. To the left of and below the asterisk are four orthochromatic megaloblasts—large cell size for maturational stage: two with delayed nuclear condensation and two with condensed nuclei with abnormal nuclear margins showing small or large budding nuclei. To the right of the asterisk are two giant band neutrophils. On the right at midfield is a plasma cell below which is a lymphocyte. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
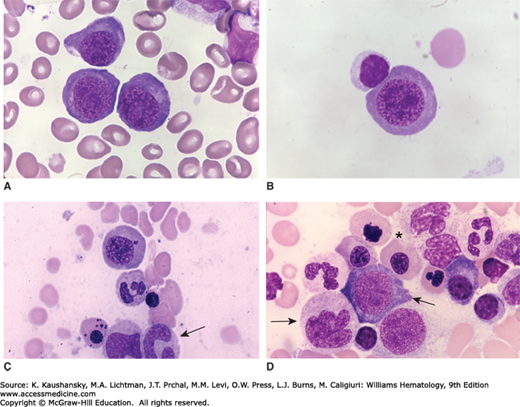
Table 41–4 lists the causes of megaloblastic anemia. By far the most common causes worldwide are folate deficiency and cobalamin deficiency. There has, however, been a marked reduction in the prevalence of folate deficiency in North America and a growing number of other countries that have implemented folic acid fortification of the food supply.
|
Megaloblastic cells have much more cytoplasm and RNA than do their normal counterparts, but they have a relatively normal amount of DNA,144 suggesting that cytoplasmic constituents (RNA and protein) are synthesized faster than is DNA. Evidence that maturation is retarded in megaloblastic precursors supports this conclusion.145 DNA synthesis is impaired,146 and migration of the DNA replication fork and the joining of DNA fragments synthesized from the lagging strand (Okazaki fragments) are delayed,147 and the S-phase is prolonged.146
Slowing of DNA replication in the megaloblastic anemias of folate and cobalamin deficiency appears to arise from failure of the folate-dependent conversion of dUMP to dTMP. Because of this failure, deoxyuridine triphosphate (dUTP) levels become abundant and because DNA polymerase is promiscuous with respect to its substrate specificity, allows dUTP to become incorporated into the DNA of folate-deficient cells in place of deoxythymidine triphosphate (dTTP).148 DNA excision–repair mechanisms to repair the DNA by replacing uridine with thymidine fail for the same reason that uridine triphosphate was incorporated into the DNA in the first place. The result is a repetitive iteration of flawed DNA repair that ultimately leads to DNA strand breaks, fragmentation, and apoptotic cell death.149
Addition of deoxyuridine (dU) to marrow cells in culture normally decreases the incorporation of tritiated thymidine into DNA, because the dU is converted via dUMP→dTMP to unlabeled dTTP, which competes with the tritiated thymidine. In megaloblastic cells, this effect of added dU is greatly diminished. This finding is consistent with impairment in the dUMP→dTMP reaction in the megaloblastic cells and was the basis for the now defunct dU suppression test.150 The failed excision–repair model following dUTP misincorporation into DNA also explains the chromosome breaks and other abnormalities that occur in megaloblastic cells.151
All megaloblastic anemias share certain general clinical features. Because the anemia develops slowly, with opportunity for cardiopulmonary and intraerythrocytic compensatory changes,152 it produces few symptoms until the hematocrit is severely depressed. Symptoms, when they appear, are those of anemia: weakness, palpitation, fatigue, light-headedness, and shortness of breath. The blending of severe pallor and slight jaundice caused by a combination of intramedullary and extravascular hemolysis produce a characteristic lemon-yellow skin. Leukocyte and platelet counts may be low, but rarely cause clinical problems. Details of the clinical manifestations are given in the sections on the specific forms of megaloblastic anemia later in this chapter.
All cell lines are affected. Erythrocytes vary markedly in size and shape, often are large and oval, and in severe cases can show basophilic stippling and nuclear remnants (Cabot rings and Howell-Jolly bodies). Erythroid activity in the marrow is enhanced, although the megaloblastic cells usually die before they are released, accounting for the reduced reticulocyte count. The more severe the anemia, the more pronounced the morphologic changes in the red cells. When the hematocrit is less than 20 percent, erythroblasts with megaloblastic nuclei, including an occasional promegaloblast, may appear in the blood. The anemia is macrocytic (mean corpuscular volume [MCV] is 100 to 150 fL or more), although coexisting iron deficiency, thalassemia trait,153 or inflammation can prevent macrocytosis.154 Slight macrocytosis may be the earliest sign of megaloblastic anemia. Because of the progressive nature of gradual replacement of normocytic red cells with the macrocytic progeny of a megaloblastic marrow, the earliest observable change in red cell indices is an increase in the red cell distribution width (RDW), reflecting an increase in anisocytosis.
Neutrophil nuclei often have more than the usual three to five lobes (see Fig. 41–12).155 Typically, more than 5 percent of the neutrophils have five lobes. Cells may contain six or more lobes, a morphology rarely seen in normal neutrophils but not pathognomonic of megaloblastic hematopoiesis. In nutritional megaloblastic anemias caused by folate deficiency, hypersegmented neutrophils are an early sign of megaloblastosis5 and persist in the blood for many days after treatment.155 Neutrophil hypersegmentation was not found to be a sensitive test for mild cobalamin deficiency.156 Cytogenetic studies are nonspecific and show chromosomes that are elongated and broken. Specific therapy corrects these abnormalities, usually within 2 days, although some abnormalities do not disappear for months.151,157 Platelets are often reduced in number and slightly smaller than normal with a wider variation in size (increased platelet distribution width [PDW]).158 The morphologic features of megaloblastic anemia may be grossly exaggerated in patients who have been splenectomized or lack a functional spleen as occurs in celiac disease or sickle cell anemia. Numerous circulating megaloblasts and bizarre red cell morphology may be present.159
Aspirated marrow is cellular and shows striking megaloblastic changes, especially in the erythroid series with well-hemoglobinized erythroblasts containing nuclei that possess less-mature, more-open nuclear chromatin than their normal counterparts. There is a preponderance of earlier basophilic erythroblasts over more mature forms which gives the overall impression of a maturation arrest (see Fig. 41–13). Sideroblasts are increased in number and contain increased numbers of iron granules. The ratio of myeloid to erythroid precursors falls to 1:1 or lower, and granulocyte reserves may be decreased. In severe cases, promegaloblasts containing an unusually large number of mitotic figures are plentiful. Macrophage iron content often is increased. Megaloblastic features in the granulocytic series is also usually present with giant forms and large horseshoe-shaped nuclei. Occasionally megakaryocytes with hyperlobated nuclei are present.
Many features of megaloblastic anemia may be masked when megaloblastic anemia is combined with a microcytic anemia.154 The anemia can be normocytic or even microcytic, whereas the blood film may show both microcytes and macroovalocytes (a “dimorphic anemia”). The marrow may contain “intermediate” megaloblasts160 that are smaller and look less “megaloblastic” than usual. In this kind of mixed anemia, the microcytic component usually is iron-deficiency anemia,154 but it may be thalassemia minor153 or the anemia of chronic disease. Even megaloblastic anemia masked by a severe microcytic anemia usually shows hypersegmented neutrophils in the blood and giant metamyelocytes and bands in the marrow. Neutrophil myeloperoxidase levels are high.161
Less commonly, the megaloblastic component of a mixed iron-deficiency anemia can be overlooked, and the patient may be treated only with iron. In this situation, the anemia may respond only partly to therapy, and megaloblastic features become more conspicuous as iron stores fill. The masking of macrocytosis in these situations may be responsible for delay or difficulty in diagnosis of pernicious anemia, particularly in certain geographic areas and ethnic groups where there is a high incidence of thalassemia and microcytic hemoglobinopathies.153,162,163 There are several situations that favor the coexistence of a megaloblastic state with iron deficiency. Both folate and iron deficiency occur in celiac disease,164 and cobalamin and iron deficiency both complicate gastric reduction surgery for morbid obesity.165 Furthermore, Helicobacter pylori infection is associated with gastric atrophy that can result first in iron deficiency and later lead to cobalamin malabsorption and perhaps even predispose to pernicious anemia.166,167
If a patient with a full-blown megaloblastic anemia receives cobalamin or folate before marrow aspiration, the anemia persists but the megaloblastic changes may be obscured. Attenuated megaloblastic changes also are seen in patients with early megaloblastic anemia, in patients with coexisting infection,154 or in patients after transfusion.
Occasionally, very severe megaloblastic anemia produces marrow morphology so bizarre as to be mistaken for acute leukemia. In some cases, the erythroid series does not mature, and the megaloblastic pronormoblast dominates the marrow with prominent mitotic figures and dysmorphic forms, raising the possibility of erythroid leukemia.
In most forms of megaloblastic anemia, cytologic abnormalities resembling megaloblastosis may appear in other proliferating cells. Epithelial cells from the mouth, stomach, small intestine, and cervix uteri may look megaloblastic, appearing larger than their normal counterparts and containing atypical immature-looking nuclei. Distinguishing these “megaloblastic” changes from the changes of malignancy may be difficult.168
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


