INTRODUCTION
SUMMARY
Red cells possess active metabolic machinery that provides energy to pump ions against electrochemical gradients, to maintain red cell shape, to keep hemoglobin iron in the reduced form, and to maintain enzyme and hemoglobin sulfhydryl groups. The main source of metabolic energy comes from glucose. Glucose is metabolized through the glycolytic pathway and through the hexose monophosphate shunt. Glycolysis catabolizes glucose to pyruvate and lactate, which represent the end products of glucose metabolism in the erythrocyte, because it lacks the mitochondria required for further oxidation of pyruvate. Adenosine diphosphate (ADP) is phosphorylated to ATP, and nicotinamide adenine dinucleotide (NAD)+ is reduced to NADH in glycolysis. 2,3-Bisphosphoglycerate, an important regulator of the oxygen affinity of hemoglobin, is generated during glycolysis. The hexose monophosphate shunt oxidizes glucose-6-phosphate, reducing NADP+ to reduced nicotinamide adenine dinucleotide phosphate (NADPH). In addition to glucose, the red cell has the capacity to utilize some other sugars and nucleosides as a source of energy. The red cell lacks the capacity for de novo purine synthesis, but has a salvage pathway that permits synthesis of purine nucleotides from purine bases. The red cell contains high concentrations of glutathione, which is maintained almost entirely in the reduced state by NADPH through the catalytic activity of glutathione reductase. Glutathione is synthesized from glycine, cysteine, and glutamic acid in a two-step process that requires ATP as a source of energy. Catalase and glutathione peroxidase serve to protect the red cell from oxidative damage. The maturation of reticulocytes into erythrocytes is associated with a rapid decrease in the activity of several enzymes. However, the decrease in activities of other enzymes occurs much more slowly or not at all with aging.
Erythrocyte enzyme deficiencies may lead to hemolytic anemia; expression of the defect in other cell lines may lead to pathologic changes such as neuromuscular abnormalities. Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common erythrocyte enzyme defect. In some populations, more than 20 percent of people may be affected by this enzyme deficiency. In the common polymorphic forms, such as G6PD A–, G6PD Mediterranean, or G6PD Canton, hemolysis occurs only during the stress imposed by infection or administration of “oxidative” drugs, and in some individuals upon ingestion of fava beans. Neonatal icterus, which appears largely with the interaction with an independent defect in bilirubin conjugation, is the clinically most serious complication of G6PD deficiency. Patients with uncommon, functionally very severe, genetic variants of G6PD experience chronic hemolysis, a disorder designated hereditary nonspherocytic hemolytic anemia.
Hereditary nonspherocytic hemolytic anemia (HNSHA) also occurs as a consequence of other enzyme deficiencies, the most common of which is pyruvate kinase (PK) deficiency. Glucose phosphate isomerase (GPI), triosephosphate isomerase (TPI), and pyrimidine 5′-nucleotidase (P5′N) deficiency are included among the relatively rare causes of hereditary nonspherocytic hemolytic anemia. In the case of some deficiencies, notably those of glutathione synthetase (GS), TPI, and phosphoglycerate kinase (PGK), the defect is expressed throughout the body, and neurologic and other defects may be a prominent part of the clinical syndrome.
Diagnosis is best achieved by determining red cell enzyme activity either with a quantitative assay or a screening test. Except for the basophilic stippling of erythrocytes that is characteristic, but not specific, of pyrimidine 5′-nucleotidase deficiency, red cell morphology is of little or no help in differentiating one red cell enzyme deficiency from another. A variety of molecular lesions have been defined in most of these enzyme deficiencies. Confirmation of the diagnosis by DNA analysis is recommended: it is necessary for genetic counseling and is helpful in recommendations for treatment, as patients with some enzyme deficiencies (e.g., GPI deficiency) tend to respond favorably to splenectomy whereas others do not (e.g., G6PD deficiency). Some of the defects, such as PK and GPI deficiencies, are transmitted as autosomal recessive disorders, whereas G6PD and PGK deficiencies are X linked.
Acronyms and Abbreviations:
ADA, adenosine deaminase; ADP, adenosine diphosphate; AK, adenylate kinase; AP-1, a transcription factor; 2,3-BPG, 2,3-bisphosphoglycerate; BPGM, bisphosphoglycerate mutase enzyme; CDP, cytidine diphosphate; 2,3-DPG, 2,3-diphosphoglycerate; EMP, Embden-Meyerhof direct glycolytic pathway; FAD, flavin adenine dinucleotide; G6PD, glucose-6-phosphate dehydrogenase; GAPDH, glyceraldehyde phosphate dehydrogenase; GCL, glutamate cysteine ligase; GLUT1, glucose transporter 1; GPI, glucose phosphate isomerase; GR, glutathione reductase; GS, glutathione synthetase; GSH, reduced glutathione; GSSG, oxidized glutathione; HFE, the gene associated with hereditary hemochromatosis; HK, hexokinase; KLF1, key erythroid transcription factor; LDH, lactate dehydrogenase; miRNA, microRNA; MRP1, multidrug resistance protein 1; NAD, nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate (reduced form); nt, nucleotide; P5′N1, pyrimidine-5′-nucleotidase-1; PFK, phosphofructose kinase; PFKM, gene encoding muscle subunit of PFK; PGK, phosphoglycerate kinase; PK, pyruvate kinase; PKLR, gene encoding PK enzyme activity in red cells and liver; SNP, single nucleotide polymorphism; SOD1, superoxide dismutase type 1; TPI, triosephosphate isomerase; WHO, World Health Organization.
DEFINITION AND HISTORY
Deficiencies in the activities of a number of erythrocyte enzymes may lead to shortening of the red cell life span. Glucose-6-phosphate dehydrogenase (G6PD) deficiency was the first of these to be recognized and is the most common.
The recognition of G6PD deficiency was the result of investigations of the hemolytic effect of the antimalarial drug primaquine, carried out in the 1950s and described in detail elsewhere.1,2,3 These early studies defined G6PD deficiency as a hereditary sex-linked enzyme deficiency that affected primarily the erythrocytes, older cells being more severely affected than newly formed ones because of age-dependent decline of mutant enzyme activity. They showed that this enzyme deficiency was very prevalent in individuals of African, Mediterranean, and Asian ethnic origins, but that it could be found in virtually any population. The common (polymorphic) forms of G6PD deficiency were found to be associated with anemia only under conditions of stress, such as the administration of oxidative drugs, infection, and the neonatal period.
Chronic hemolysis in the absence of a stress occurs in uncommon, functionally severe forms of G6PD deficiency and in patients with a variety of other red cell enzyme deficiencies. Such patients suffer from hereditary nonspherocytic hemolytic anemia. Although patients fitting the description of hereditary nonspherocytic hemolytic anemia had been documented earlier, the designation was first introduced by Crosby in 1950.4 Dacie and colleagues5 subsequently reported several families in which affected members manifested hemolytic anemia from an early age and in whom the osmotic fragility of the red cells was normal. The latter finding was the main feature that distinguished this disorder from hereditary spherocytosis. Thus, defined essentially by exclusion as a hereditary hemolytic anemia that is not hereditary spherocytosis (or without any other major aberration of red cell morphology), it is not at all surprising that hereditary nonspherocytic hemolytic anemia has proven to be extremely heterogeneous both in etiology and in clinical manifestations. Sometimes this disorder is also designated congenital nonspherocytic hemolytic anemia, but the name hereditary hemolytic anemia is more accurate and is therefore preferable. Although hereditary ovalocytosis, pyropoikilocytosis, and stomatocytosis (Chap. 46), and even thalassemia major and sickle cell disease (Chaps. 48 and 49), are hereditary hemolytic anemias that are also nonspherocytic, they are not included in this category.
Although a deficiency of G6PD was found to be responsible for hemolysis in a few patients with hereditary nonspherocytic hemolytic anemia, in the overwhelming majority of cases the cause remained obscure. In 1954, Selwyn and Dacie6 studied autohemolysis (spontaneous lysis of red cells after sterile incubation for 24 to 48 hours at 37°C) in four patients with hereditary nonspherocytic hemolytic anemia and found that in two of them lysis was only slightly increased and was prevented by glucose; these patients were designated as type 1, whereas the others, in whom glucose failed to correct autohemolysis, were classified as type 2. Autohemolysis of the erythrocytes of type 2 patients was modified by the addition of ATP. However, ATP does not penetrate the red cell membrane and instead, its modifying influence was nonspecific exerted chiefly by virtue of its effect on the osmolarity and pH of the suspending solution. These findings suggested to DeGruchy and associates7 that patients with type 2 autohemolysis suffered from a defect in ATP generation. This proposal, born of a misunderstanding of red cell biochemistry, turned out to be correct, as one of the major causes of hereditary nonspherocytic hemolytic anemia proved to be a deficiency of the ATP-generating enzyme pyruvate kinase (PK).8 PK deficiency was the first of a large number of enzyme defects that have been shown to account for this heterogeneous syndrome.
EPIDEMIOLOGY
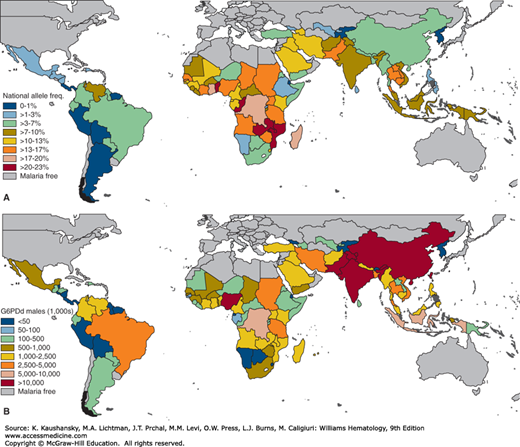
The most common red cell enzyme abnormality is deficiency of G6PD. Its prevalence among white populations ranges from less than 1 in 1000 among northern European populations to 50 percent of the males among Kurdish Jews. The lowest frequencies of G6PD deficiency are found in both North and South America (≤1 percent), and highest rates are predicted across the tropical belt of sub-Saharan Africa (15 to 30 percent; Fig. 47–1). The distribution across Asia and Asia Pacific is generally heterogeneous, ranging from virtually absent to relatively high.9,10 Although many of the highest frequencies are predicted from sub-Saharan African countries, the very high population densities across Asia infers that the overall population burden is largely focused here. The overall allele frequency of G6PD deficiency across all malaria endemic countries is predicted to be 8 percent. This corresponds to 220 million affected males and an even greater number of females, although some heterozygous females for this X-chromosome–encoded gene do not have enough deficient erythrocytes to become prone to significant hemolysis.9 Details on the distribution of G6PD deficiency among various population groups is presented elsewhere. 9,10,11,12
Figure 47–1.
Estimated prevalence of glucose-6-phosphate dehydrogenase (G6PD) deficiency. A. National-level allele frequencies. B. National-level population estimates of G6PD-deficient (G6PDd) males. (Reproduced with permission from Howes RE, Piel FB, Patil AP, et al: G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: a geostatistical model-based map. PLoS Med 2012;9(11):e1001339.)
The high frequency of G6PD-deficient genes in many populations implies that G6PD deficiency confers a selective advantage. The suggestion that resistance to malaria accounts for the high frequency of G6PD deficiency paralleling the worldwide distribution of malaria is supported by the sheer diversity of variants in the G6PD gene. Many of these are found at polymorphic frequencies in genetically isolated populations, suggesting independent selection of each variant.11,12,13 Important supporting evidence was obtained from studies in heterozygotes for G6PD A– that showed a higher degree of infestation of G6PD-sufficient cells than of G6PD-deficient cells.14 Deficient cells infested with malaria parasites may be phagocytosed more efficiently than normal cells.15 Which G6PD genotypes confer protection from malarial infection is the subject of debate.16,17,18 The majority of studies conclude that G6PD deficiency in hemizygous males, and probably also homozygous females, confers significant protection against malarial infection. The nature of protection from the mosaic state of G6PD deficiency in heterozygous females remains to be established.18,19
It has been suggested that a higher prevalence of G6PD deficiency in individuals with sickle cell disease than in the general African population reflects a favorable effect of the enzyme deficiency on the clinical course of the sickling disorders.20 However, studies aimed at investigating the effects of G6PD deficiency on the clinical manifestations of sickle cell disease have produced conflicting results. Some studies found no evidence for any such effect,21,22 whereas others reported on lower hemoglobin levels in patients with both disorders, either accompanied23 or not24 by signs of increased hemolysis.
PK deficiency is the most common cause of hereditary nonspherocytic hemolytic anemia. Based on large-scale mutation analysis, it has been estimated that the population prevalence of PK deficiency among whites is approximately 50 cases per 1 million population.25 Estimates of frequencies of other deficiency alleles, such as those for adenylate kinase, diphosphoglycerate mutase, enolase, triosephosphate isomerase (TPI), and phosphoglycerate kinase (PGK), have been made on large numbers of cord bloods.26 A particularly high incidence of heterozygous TPI deficiency (>4 percent) in Americans of African descent is supported by family studies.27 Because this is not reflected in a correspondingly high birth incidence, the allele might be lethal in the homozygous state.
In addition to the common G6PD mutations, there are mutations in other enzymes that are repeatedly encountered in the population. In PK, the c.1529G>A, p.(Arg510Gln) mutation is the most common mutation in the United States,28 and in northern and central Europe29; the c.1456C>T, p.(Arg486Trp) mutation is prevalent in southern Europe,30 and the c.1468C>T, (p.Arg490Trp) mutation in Asia.31 Similarly, the c.315G>C, p.(Glu104Asp) mutation is recurrently encountered in TPI.32 In phosphofructose kinase (PFK) deficiency, one-third of the reported patients are of Jewish origin, and in this population, an intronic splice site mutation, c.237+1G>A, and a single base-pair deletion, c.2003delC, are among the most frequently encountered mutations.33 In a number of these instances, the existence of each mutation in the context of the same haplotype implies that there has been a founder effect, that is, the mutation occurred only once, and all individuals now carrying it are descendants of the person who sustained the original mutation. The expansion of the mutation could represent a selective advantage for heterozygotes, but also may result from random factors or from a selective advantage provided by one or more tightly linked genes.
ETIOLOGY AND PATHOGENESIS
Although the binding, transport, and delivery of oxygen do not require the expenditure of metabolic energy by the red cell, a source of energy is required if the red cell is to perform its function efficiently and to survive in the circulation for its full life span of approximately 120 days. This energy is needed to maintain (1) the iron of hemoglobin in the divalent form; (2) the high potassium and low calcium and sodium levels within the cell against a gradient imposed by the high plasma calcium and sodium and low plasma potassium levels; (3) the sulfhydryl groups of red cell enzymes, hemoglobin, and membranes in the active, reduced form; and (4) the biconcave shape of the cell. If the red cell is deprived of a source of energy, it becomes sodium and calcium logged and potassium depleted, and the red cell shape changes from a flexible biconcave disk. Such a cell is quickly removed from the circulation by the filtering action of the spleen and the monocyte–macrophage system. Even if it survived, such an energy-deprived cell would gradually turn brown as hemoglobin is oxidized to methemoglobin by the very high concentrations of oxygen within the erythrocyte and would then be unable to perform its function of transporting oxygen and carbon dioxide.
The process of extracting energy from a substrate, such as glucose, and of utilizing this energy is carried out by a large number of enzymes (Table 47–1). Because the red cell loses its nucleus before it enters the circulation and most of its RNA within 1 or 2 days of its release into the circulation, it does not have the capacity to synthesize new proteins to replace those that may become degraded during its life span. The enzymes present in the red cells were formed largely by the nucleated cell in the marrow and, to a lesser extent, the reticulocyte.
| Enzyme | Activity at 37°C IU/g Hgb (mean ± SD) |
|---|---|
| Acetylcholinesterase | 36.93 ± 3.83 |
| Adenosine deaminase | 1.11 ± 0.23 |
| Adenylate kinase | 258 ± 29.3 |
| Aldolase | 3.19 ± 0.86 |
| Bisphosphoglyceromutase | 4.78 ± 0.65 |
| Catalase | 153,117 ± 2390 |
| Enolase | 5.39 ± 0.83 |
| Galactokinase | 0.0291 ± 0.004 |
| Galactose-4-epimerase | 0.231 ± 0.061 |
| Glucose phosphate isomerase | 60.8 ± 11.0 |
| Glucose-6-phosphate dehydrogenase | 8.34 ± 1.59 |
| γ-Glutamylcysteine synthetase | 1.05 ± 0.19 |
| Glutathione peroxidase* | 30.82 ± 4.65 |
| Glutathione reductase without FAD | 7.18 ± 1.09 |
| Glutathione reductase with FAD | 10.4 ± 1.50 |
| Glutathione-S-transferase | 6.66 ± 1.81 |
| Glutathione synthetase | 0.34 ± 0.06 |
| Glyceraldehyde phosphate dehydrogenase | 226 ± 41.9 |
| Hexokinase | 1.78 ± 0.38 |
| Lactate dehydrogenase | 200 ± 26.5 |
| Monophosphoglyceromutase | 37.71 ± 5.56 |
| NADH-methemoglobin reductase | 19.2 ± 3.85(30°) |
| NADPH diaphorase | 2.26 ± 0.16 |
| Nucleoside phosphorylase | 359 ± 32 |
| Phosphofructokinase | 11.01 ± 2.33 |
| Phosphoglucomutase | 5.50 ± 0.62 |
| Phosphoglycerate kinase | 320 ± 36.1 |
| Phosphoglycolate phosphatase | 1.23 ± 0.10 |
| Phosphomannose isomerase | 0.054 ± 0.026 |
| Pyrimidine 5′-nucleotidase | 0.138 ± 0.018 |
| Pyruvate kinase | 15.0 ± 1.99 |
| 6-Phosphogluconate dehydrogenase | 8.78 ± 0.78 |
| 6-Phosphogluconolactonase | 50.6 ± 5.9 |
| Ribosephosphate isomerase | 200 |
| Superoxide dismutase | 2225 ± 303 |
| Transaldolase | 1.21 ± 0.24 |
| Transketolase | 0.725 ± 0.17 |
| Triose phosphate isomerase | 2111 ± 397 |
Glucose is the normal energy source of the red cell. It is metabolized by the erythrocyte along two major routes: the glycolytic pathway and the hexose monophosphate shunt. The steps in these pathways are essentially the same as those found in other tissues and in other organisms, including even relatively simple ones such as Escherichia coli and yeast. Unlike most other cells, however, the red cell lacks mitochondria and hence a citric acid cycle. Only the reticulocytes maintain some capacity for the breakdown of pyruvate to CO2, with the attendant highly efficient production of ATP. The mature red cell extracts energy from glucose almost solely by anaerobic glycolysis. Before glucose can be metabolized by the red cell, it must pass through the membrane. Transport into the interior of the cell is facilitated by glucose transporter 1 receptor GLUT1, and perhaps regulated by the abundantly expressed membrane protein stomatin; however, the function of this protein is not fully defined.34 In humans, and other mammals that have lost the ability to synthesize ascorbic acid from glucose, GLUT1 also facilitates transport of L-dehydroascorbic acid.34 The red cell membrane contains insulin receptors, but the transport of glucose into red cells is independent of insulin.
Direct glycolytic pathway. In the Embden-Meyerhof direct glycolytic pathway (EMP; Fig. 47–2), glucose is catabolized anaerobically to pyruvate or lactate. Although 2 moles of high-energy phosphate in the form of ATP are used in preparing glucose for its further metabolism, up to 4 moles of adenosine diphosphate (ADP) may be phosphorylated to ATP during the metabolism of each mole of glucose, giving a net yield of 2 moles of ATP per mole of glucose metabolized. The rate of glucose utilization is limited largely by the hexokinase and PFK reactions. Both of the enzymes catalyzing these reactions have a relatively high pH optimum and have very little activity at pH levels lower than 7. For this reason, red cell glycolysis is very pH sensitive, being stimulated by a rise in pH. However, at higher than physiologic pH levels, the stimulation of hexokinase and phosphofructokinase activity merely results in the accumulation of fructose diphosphate and triosephosphates, because the availability of nicotinamide adenine dinucleotide (NAD)+ for the glyceraldehyde phosphate dehydrogenase (GAPDH) reaction becomes a limiting factor.
Figure 47–2.
Glucose metabolism of the erythrocyte. The details of the hexose monophosphate pathway are shown in Fig. 47–3.
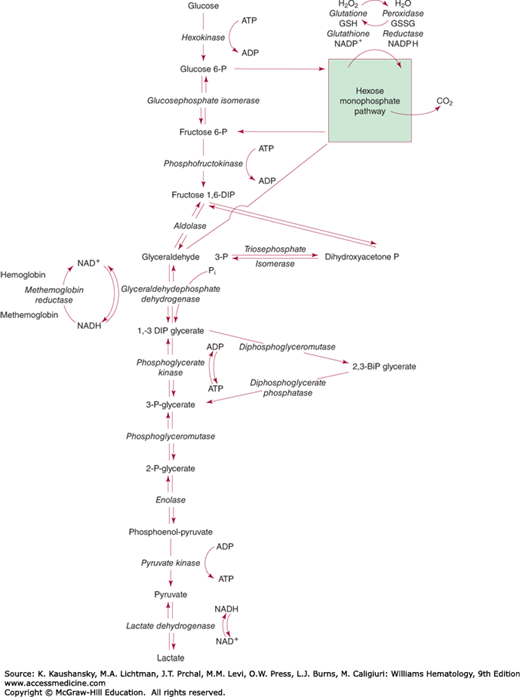
Branching of the metabolic stream after the formation of 1,3-bisphosphoglycerate (1,3-BPG) provides the red cell with flexibility in regard to the amount of ATP formed in the metabolism of each mole of glucose. 1,3-BPG may be metabolized to 2,3-bisphosphoglycerate (2,3-BPG), also known as 2,3-diphosphoglycerate (2,3-DPG), thus “wasting” the high-energy phosphate bond in position 1 of the glycerate. Removing the phosphate group at position 2 by bisphosphoglycerate phosphatase results in the formation of 3-phosphoglycerate. Both reactions in this unique glycolytic bypass, known as the Rapoport-Luebering shunt, are catalyzed by the erythroid-specific multifunctional enzyme bisphosphoglycerate mutase.35 In mammalian erythrocytes, a separate 2,3-BPG phosphatase activity has been ascribed to multiple inositol polyphosphate phosphatase.36 In contrast to bisphosphoglycerate mutase, multiple inositol polyphosphate phosphatase-1 is able to remove the phosphate at position 3, thereby bypassing the formation of 3-phosphoglycerate. The precise functional significance of multiple inositol polyphosphate phosphatase-1 for human red cell physiology and regulation of 2,3-BPG levels remains to be established.
3-Phosphoglycerate may also be formed directly from 1,3-BPG through the PGK step, resulting in phosphorylation of 1 mole of ADP to ATP. Although metabolism of glucose through the 2,3-BPG step occurs without any net gain of high-energy phosphate bonds in the form of ATP, metabolism through the PGK step results in the formation of two such bonds per mole of glucose metabolized. This portion of the direct glycolytic pathway has been called the energy clutch. Regulation of metabolism at this branch point determines not only the rate of ADP phosphorylation to ATP but also the concentration of 2,3-BPG, an important regulator of the oxygen affinity of hemoglobin (Chaps. 49 and 57). The concentration of 2,3-BPG depends on the balance between its rate of formation and degradation by bisphosphoglycerate mutase. Hydrogen ions inhibit the bisphosphoglycerate mutase reaction and stimulate the phosphatase reaction. Thus, red cell 2,3-BPG levels are exquisitely sensitive to pH: a rise in pH causes a rise in 2,3-BPG levels, whereas acidosis results in 2,3-BPG depletion. It may be that the ratio of oxyhemoglobin to deoxyhemoglobin also influences 2,3-BPG synthesis by virtue of the fact that only deoxyhemoglobin binds this compound, thus affecting the concentration of free 2,3-BPG that is available for feedback inhibition of the enzymes that lead to its formation. However, the available evidence suggests that the pH is the primary controlling factor.
Metabolism of glucose by way of the EMP may also yield reducing energy in the form of the reduced form of NAD (NADH). The reduction of NAD+ to NADH occurs in the GAPDH step. If NADH is reoxidized in reducing methemoglobin to hemoglobin, the end product of glucose metabolism is pyruvate. If NADH is not reoxidized by methemoglobin, however, pyruvate is reduced in the lactate dehydrogenase (LDH) step, forming lactate as the final end product of glucose metabolism. The lactate or pyruvate formed is transported from the red cell and is metabolized elsewhere in the body. Thus, the erythrocyte has a flexible EMP that can adjust the amount of ADP phosphorylated per mole of glucose according to the requirement of the cell.
The regulation of red cell glycolytic metabolism is very complex. Products of some reactions may stimulate others. For example, the PK reaction is exquisitely sensitive to fructose 1,6-diphosphate, the product of PFK. Conversely, other metabolic products may serve as strong enzyme inhibitors. In addition, there is increasing evidence that glycolytic enzymes assemble into enzyme complexes to the interior of the red cell membrane.37 The assembly of these complexes seems to be regulated by the oxygen status of hemoglobin and the phosphorylation status of band 3,37,38 suggesting they play a direct role in the regulation of oxygen-dependent changes in glycolytic and pentose shunt fluxes.39
Notably, a number of glycolytic enzymes show additional functional activities. For instance, in addition to its role in glycolysis, glucosephosphate isomerase also functions as a neuroleukin or autocrine motility factor. Another example is enolase, that has been reported to also function as plasminogen receptor.40,41 The additional functional activities of these “moonlighting” enzymes could contribute to the complexity of the phenotype of the associated disorder.
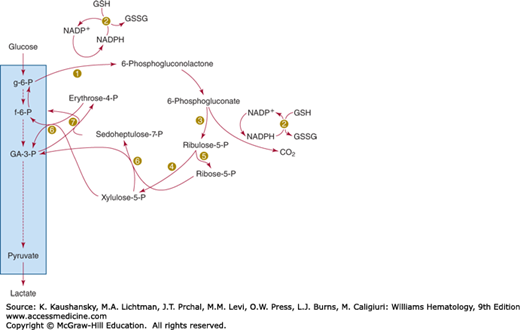
Not all the glucose metabolized by the red cell passes through the direct glycolytic pathway. A direct oxidative pathway of metabolism, the hexose monophosphate shunt, also functions. In this pathway, glucose-6-phosphate is oxidized at position 1, yielding carbon dioxide. In the process of glucose oxidation, NADP+ is reduced to NADPH (reduced NAD phosphate). The pentose phosphate formed when glucose is decarboxylated undergoes a series of molecular rearrangements, eventuating in the formation of a triose, glyceraldehyde-3-phosphate, and a hexose, fructose-6-phosphate (Fig. 47–3). These are normal intermediates in anaerobic glycolysis and thus can rejoin that metabolic stream. Because the glucose phosphate isomerase reaction is freely reversible, allowing fructose-6-phosphate to be converted to glucose-6-phosphate, recycling through the hexose monophosphate pathway is also possible. Unlike the anaerobic glycolytic pathway, the hexose monophosphate pathway does not generate any high-energy phosphate bonds. Its primary function appears to be the formation of NADPH, and, indeed, the amount of glucose passing through this pathway appears to be regulated by the amount of NADP+ that has been made available by the oxidation of NADPH. NADPH appears to function primarily as a substrate for the reduction of glutathione-containing disulfides in the erythrocyte through mediation of the enzyme glutathione reductase, which catalyzes the conversion of oxidized glutathione (GSSG) to reduced glutathione (GSH) and the reduction of mixed disulfides of hemoglobin and GSH.42
Hexokinase catalyzes the phosphorylation of glucose in position 6 by ATP. It thus serves as the first step in the utilization of glucose, whether by the anaerobic or the hexose monophosphate pathway. Mannose or fructose may also serve as a substrate for this enzyme. Hexokinase is the glycolytic enzyme with the lowest activity. Reticulocytes have much higher levels of hexokinase activity than do mature red cells.43,44
Hexokinase has an absolute requirement for magnesium. It is strongly inhibited by its product, glucose-6-phosphate, and is released from this inhibition by the inorganic phosphate ion45,46 and by high concentrations of glucose.47 Inorganic phosphate enhances the rate of glucose utilization by red cells. This effect is not exerted through hexokinase but through stimulation of the PFK reaction, resulting in a lowered glucose-6-phosphate concentration within the cell and thus releasing hexokinase from inhibition.48 GSSG49 and other disulfides, as well as 2,3-BPG,50 inhibit hexokinase. The determination of the structures of the human and rat hexokinase isozymes have provided substantial insight into ligand-binding sites and subsequent modes of interaction of these ligands.51,52
The two major fractions of red cell hexokinase (HK) have been designated HKI and HKR; the latter fraction being unique to erythrocytes and particularly to reticulocytes.53 Both red cell isozymes are monomers and produced from the hexokinase I gene (HK1),54 with an apparent molecular weight of 112 kDa.55 The HK1 gene is localized on chromosome 10q22 and spans more than 100 kb. It contains 29 exons,56,57 which, by tissue-specific transcription, generate multiple transcripts by alternative use of the 5′ exons.57,58 Erythroid-specific transcriptional control results in a unique red blood cell–specific mRNA that differs from HKI transcripts at the 5′ end. Consequently, HKR lacks the porin-binding domain that mediates HKI binding to mitochondria.59 A single nucleotide polymorphism (SNP) in the first intron of HKR was found to be strongly associated with reduced hemoglobin and hematocrit levels in the European population.60 HK deficiency is a rare cause of hereditary nonspherocytic hemolytic anemia.
Glucose phosphate isomerase (GPI) catalyzes the interconversion of glucose-6-phosphate and fructose-6-phosphate, the second step of the EMP. The crystal structure of human GPI has been resolved. The enzyme is a homodimer, composed of two subunits of 63 kDa each. The enzyme’s active site is composed of polypeptide chains from both subunits, making the dimeric form essential for catalytic activity.61 Residues that are not in direct contact with the reacting substrate molecule have also been implicated as important for catalytic function of GPI.62 The gene encoding GPI (GPI) is located on chromosome 19q13.1 and consists of 18 exons, spanning at least 50 kb, with a complementary DNA (cDNA) of 1.9 kb in length.63 GPI deficiency is one of the relatively more common causes of hereditary nonspherocytic hemolytic anemia.
PFK catalyzes the rate-limiting phosphorylation of fructose-6-phosphate by ATP to fructose-1,6-diphosphate. Under intracellular conditions this reaction is nearly irreversible. Therefore PFK is an important regulator of glycolytic flux. The enzyme has a molecular mass of around 340 kDa. Red cell phosphofructokinase exists as five different homo- or heterotetramers comprised of muscle (M) and liver (L) subunits. Each tetramer displays unique properties with respect to catalytic function and regulation. The enzyme requires magnesium for activity. PFK activity is tightly controlled and subject to regulation by many metabolic effectors. Among the most important activators are ADP, cyclic adenosine monophosphate (cAMP), and fructose-2,6-diphosphate, whereas PFK is inhibited by its substrate ATP, citrate, and lactate.64,65 These metabolic effectors exert their effects probably by stabilizing either the minimally active dimeric and fully active tetrameric form of PFK.66 PFK activity is also regulated by its binding to calmodulin,67 and association of the enzyme with the red cell membrane68; in particular, binding to band 369,70 and actin71 appears to inhibit and stimulate PFK activity, respectively.
A preliminary crystallographic analysis of dimeric wild-type human muscle PFK has been presented.72 The gene encoding the 85-kDa M subunit (PFKM) is located on chromosome 12q13.3 and spans 30 kb. It contains 27 exons and at least three promoter regions.73 The 80-kDa L-subunit encoding gene (PFKL) is located on chromosome 21q22.3; it contains 22 exons and spans more than 28 kb.74 Deficiency of PFK is associated with mild hemolytic anemia and with type VII glycogen storage disease (Tarui disease).75
Aldolase reversibly cleaves fructose-1,6-diphosphate into two trioses. The “upper” half of the fructose-1,6-diphosphate molecule becomes dihydroxyacetone phosphate and the “lower” half becomes glyceraldehyde-3-phosphate. Aldolase is a 159-kDa homotetrameric enzyme comprised of subunits of 40 kDa each.76 Three distinct isoenzymes have been identified: aldolase A, B, and C. The 364-amino-acids-long aldolase A subunits are primarily expressed in erythrocytes and muscle cells.77 The structure of human aldolase A is known.78 Red cell aldolase binds to F-actin79 and the N-terminal part of band 3,80 which inhibits its activity.70 The gene for aldolase A (ALDOA) is located on chromosome 16q22–24. It spans 7.5 kb and consists of 12 exons. Several transcription-initiation sites were identified and ALDOA pre-mRNA is spliced in a tissue-specific manner.81 Aldolase deficiency is a very rare cause of hereditary nonspherocytic hemolytic anemia.
TPI is the enzyme of the anaerobic glycolytic pathway that has the highest activity. Its metabolic role is to catalyze interconversion of the two trioses formed by the action of aldolase: dihydroxyacetone phosphate and glyceraldehyde-3-phosphate.82 Although equilibrium is in favor of dihydroxyacetone phosphate, glyceraldehyde-3-phosphate undergoes continued oxidation through the action of GAPDH and is thus removed from the equilibrium. TPI is a dimer consisting of two identical 27-kDa subunits of 248 amino acids.83 Several crystal structures have been resolved.84,85 These show that the active site is at the dimer interface and a number of critical residues have been identified. Several water molecules, some of which are highly conserved, are an integral part of the dimer interface. There are no isoenzymes known, but three distinct electrophoretic forms can be distinguished as a result of posttranslational modifications.86 Red blood cell TPI activity is not red-cell-age dependent. TPI is transcribed from a single gene (TPI1), located on chromosome 12p13. The gene spans 3.5 kb and contains 7 exons. Three processed pseudogenes have been identified.87 A deficiency of TPI causes hereditary nonspherocytic hemolytic anemia and a severe neuromuscular disorder.
GAPDH performs the dual functions of oxidizing and phosphorylating glyceraldehyde-3-phosphate, producing 1,3-BPG. In the process, NAD+ is reduced to NADH. This enzyme is closely associated with the red cell membrane88 where it binds to the N-terminal part of band 3.70 Membrane binding influences the activity of GAPDH89 thereby possibly regulating glycolytic flux.90 Human red blood cell GAPDH has been purified. It is a homotetramer of approximately 150 kDa, composed of 36-kDa subunits, and shows an absolute specificity for NAD+.91 One of the many nonglycolytic functions of GAPDH40,41 may include its function as a transferrin receptor.92 The crystal structure of human liver GAPDH reveals a homotetramer, each subunit of which is bound to a NAD+ molecule.93 Deficiency of GAPDH seems a rare occurrence without functional consequences.94
PGK effects the transfer to ADP of the high-energy phosphate from the 1-carbon of 1,3-diphosphoglycerate (1,3-DPG) to form ATP. The reaction is readily reversible and can be bypassed by the Rapoport-Luebering shunt. The isoenzyme PGK-1 is ubiquitously expressed in all somatic cells and is a 48-kDa monomeric enzyme of 417 amino acids.95 Expression of isozyme PGK-2 has been found in testis.96 PGK is composed of two domains. The N-terminal domain binds 3-phosphoglycerate and 1,3-DPG, whereas ADP and ATP bind to the C-terminal domain. For catalysis to occur, the protein needs to undergo a large conformational change (“hinge bending”).97,98,99 The gene encoding PGK (PGK-1) is located on the long arm of the X-chromosome (Xq13), spans 23 kb, and is composed of 11 exons. Nonfunctional pseudogenes have been located on chromosome 19 and the X-chromosome.96 Deficiency of PGK is a rare cause of nonspherocytic hemolytic anemia, often associated with neuromuscular abnormalities.
The same protein molecule is responsible for both bisphosphoglycerate mutase and bisphosphoglycerate phosphatase activities in the erythrocyte.35,100 This enzyme is particularly important because it regulates the concentration of 2,3-BPG of erythrocytes. In its role as a bisphosphoglyceromutase, the enzyme competes with PGK for 1,3-BPG as a substrate. It changes 1,3-BPG to 2,3-BPG, thereby dissipating the energy of the high-energy acylphosphate bond.101 It is inhibited by its product 2,3-BPG and by inorganic phosphate, and it is activated by 2-phosphoglycerate and by increased pH levels. It requires 3-phosphoglycerate for activity. In its role as bisphosphoglycerate phosphatase it catalyzes the removal of the phosphate group from carbon 2 of 2,3-BPG.101 It is inhibited by its product 3-phosphoglycerate and by sulfhydryl reagents. It is most active at a slightly acid pH and is strongly stimulated by bisulfite and phosphoglycolate. Phosphoglycolate, the most potent activator of phosphatase activity, is present in erythrocytes at very low concentrations,102 but the source of this substance in red cells is unknown.103,104 Phosphoglycolate phosphatase, the enzyme that hydrolyzes phosphoglycolate, has also been identified in erythrocytes.105
Bisphosphoglycerate mutase is a homodimer, with 30-kDa subunits consisting of 258 amino acids. The crystal structure of human bisphosphoglycerate mutase has been determined, providing a rationale for the specific residues that are crucial for synthase, mutase, and phosphatase activity.106,107 The gene for bisphosphoglycerate mutase (BPGM) has been mapped to chromosome 7q31–34 and it consists of 3 exons, spanning more than 22 kb.
A deficiency of bisphosphoglycerate mutase results in a marked decrease in red cell 2,3-BPG levels. The consequent left shift of the oxygen dissociation curve leads to erythrocytosis/polycythemia (Chap. 57).
An equilibrium is established between 3-phosphoglycerate and 2-phosphoglycerate by phosphoglycerate mutase.108 2,3-BPG acts as an essential cofactor for the transformation. Red blood cell phosphoglycerate mutase is a heterodimer consisting of M and B subunits, encoded by separate genes.109 Only one well-characterized patient with partial red blood cell monophosphoglycerate mutase deficiency has been described who was homozygous for a p.Met230Ile amino acid change in the B subunit.110 The mutant enzyme was unstable and thus had a short half-life.111 Unexpectedly, all glycolytic intermediates were decreased, possibly because of lactate accumulation.112 The exact clinical consequences of this red blood cell enzymopathy remain to be established.
Enolase is a homodimeric enzyme that establishes an equilibrium between 2-phosphoglycerate and phosphoenolpyruvate. The reaction is facilitated by the presence of metal ions.113 Aside from its enzymatic function in the glycolytic pathway, α-enolase (ENO1) has been implicated in numerous diseases, including metastatic cancer, autoimmune disorders, ischemia, and bacterial infection.114 The gene for enolase (ENO1) is located on chromosome 1p36.23. Enolase deficiency is extremely rare. Although it has been reported in association with hereditary nonspherocytic hemolytic anemia,115,116 a clear cause-and-effect relationship has not yet been firmly established.
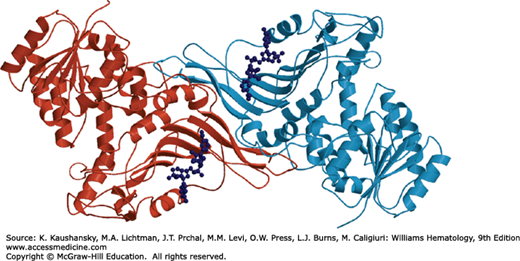
The transfer of phosphate from phosphoenolpyruvate to ADP, forming ATP and pyruvate, is catalyzed by the allosteric enzyme PK. This is the second energy-yielding step of glycolysis. Four PK isoenzymes are present in mammalian tissues: PK-M1 (in skeletal muscle), PK-M2 (in leukocytes, kidney, adipose tissue, and lungs), PK-L (in liver), and PK-R (in red blood cells). The four PK isoenzymes are products of only two genes (PKLR and PKM2). The PK-M1 and PK-M2 enzymes are formed from the PKM2 gene by alternative splicing.117 PK-L (the liver enzyme) and PK-R (the erythrocyte enzyme) are products of the PKLR gene, transcribed from two different, tissue-specific promoters.118,119 There is evidence that other, yet unknown, regulatory elements are involved in PKLR gene expression.120 PKLR consists of 12 exons and spans more than 10 kb. Exon 2, but not exon 1, is present in the processed liver transcript; in the red cell enzyme exon 1, but not exon 2, is represented.119 The red blood cell–specific mRNA is 2 kb in length and codes for a full-length 63-kDa PK-R subunit of 574 amino acids.121 Red blood cell PK is a heterotetramer comprised of two 62- to 63-kDa and two 57- to 58-kDa subunits, the latter resulting from limited proteolytic cleavage of the full-length subunit.122,123 Each subunit of PK-R contains an N domain, A domain, B domain, and C domain (Fig. 47–4).124 Domain A is the most highly conserved, whereas the B and C domains are more variable.125 The functional role of the N domain is unknown, but it may play a role in enzyme regulation.126,127 The active site of PK lies in a cleft between the A domain and the flexible B domain. The C domain contains the binding site for fructose-1,6-diphosphate. Both intra- and intersubunit interactions are considered to be key determinants of the allosteric response, which involves switching of the PK tetramer from the low-affinity T state to the high-affinity R state.128,129,130,131,132 Red cell PK manifests sigmoid kinetics with respect to phosphoenolpyruvate in the absence of fructose-1,6-diphosphate. Hyperbolic kinetics are observed in the presence of even minute amounts of fructose-1,6-diphosphate,122,133 so that at low concentrations of phosphoenolpyruvate the enzyme activity is greatly increased by fructose diphosphate. PK deficiency is the most common cause of hereditary nonspherocytic hemolytic anemia.
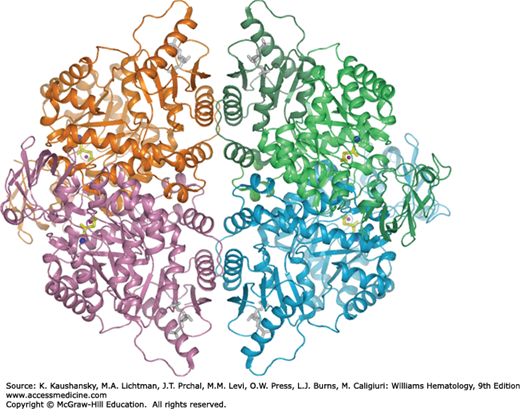
Figure 47–4.
Ribbon representation of the human erythrocyte pyruvate kinase tetramer. The substrate phosphoglycolate and fructose-1,6-diphosphate are shown in ball-and-stick representation, and colored yellow and gray, respectively. Metal ions in the active site are shown as blue (potassium) and pink (manganese) spheres. Individual subunits are colored lime, cyan, violet, and orange.
LDH catalyzes the reversible reduction of pyruvate to lactate by NADH, the last step in the EMP. The enzyme is composed of H (heart) and M (muscle) subunits. In red cells, the predominant subunit is H. Hereditary absence of the H subunit seems to be a benign condition, usually without clinical manifestations,134,135 although one case with hemolysis has been reported.136 Absence of the M subunit has been reported as well,137 and was unaccompanied by hematologic manifestations. Judging from the origin of the reports, LDH deficiency appears to be most common in Japan, where population surveys show a gene frequency of approximately 0.05 for each deficiency, and several mutations have been identified.138
G6PD is the most extensively studied erythrocyte enzyme. It catalyzes the oxidation of glucose-6-phosphate to 6-phosphogluconolactone, which is rapidly hydrolyzed to 6-phosphogluconic acid, in the first step of the hexose monophosphate pathway. NADP+ is reduced to NADPH in this reaction, generating 1 mole of NADPH. In the erythrocyte, the hexose monophosphate pathway is the only source of NADPH, which is crucial in maintaining high cellular levels of GSH to protect the cell from oxidative stress-induced damage.
The G6PD monomer is composed of 515 amino acids with a calculated molecular weight of approximately 59 kDa.139 Aggregation of these inactive monomers into catalytically active dimers and higher forms requires the presence of NADP+ (Fig. 47–5).140 Hence, NADP+ is bound to the enzyme both as a structural component, in the subunit interface, and as one of the substrates of the reaction.141,142,143 Under physiologic conditions, the active human enzyme exists in a dimer–tetramer equilibrium. Lowering the pH causes a shift toward the tetrameric form.141,144,145
G6PD is strongly inhibited by physiologic amounts of NADPH146 and, to a lesser extent, by physiologic concentrations of ATP.147 It has much higher enzyme-activity in reticulocytes than in mature red cells, especially for the mutant forms of the enzyme.43,148 This may complicate diagnosing G6PD deficiency in patients with high reticulocyte counts following a hemolytic episode (see “Laboratory Features” below).
The three-dimensional model of the crystal structure of human G6PD shows that the G6PD monomer is built up by two domains, a N-terminal domain and a large β + α domain with an antiparallel nine-stranded sheet. The extensive interface between the two monomers is of crucial importance for enzymatic stability and activity.143 The fully conserved amino acids 198 to 205 (Arg-Ile-Asp-His-Tyr-Leu-Gly-Lys) are essential for substrate binding and catalysis.143,149,150,151
The G6PD gene is located on the X-chromosome (Xq28). It spans 18 kb, and consists of 13 exons of which exon 1 is noncoding. Methylation of certain cytidines at the 3′ end is believed to have a regulatory function.152,153 The 3′-UTR (untranslated region) also harbors putative microRNA (miRNA) target sites, which potentially could have a functional effect on the downregulation of G6PD mRNA, thereby affecting the stability of G6PD mRNA and translation, or the miRNA regulation process.154 G6PD deficiency is one of the world’s most common hereditary disorders. Many mutations and variants have been reported and studied.1,2,155,156,157,158,159,160
Although 6-phosphogluconolactone is the direct product of the oxidation of glucose-6-phosphate by G6PD and hydrolyzes spontaneously at a physiologic pH, enzymatic hydrolysis is much more rapid and is required for normal metabolic flow through the stimulated hexose monophosphate pathway.161,162 Partial deficiency of the enzyme has been observed163 and is probably benign.164
Phosphogluconate dehydrogenase catalyzes the oxidation of phosphogluconate to ribulose-5-phosphate and CO2 and the reduction of NADP+ to NADPH. Variability of electrophoretic mobility of the enzyme is common in humans and in several animal species.165 Deficiency of the enzyme has been observed only rarely and appears to be essentially innocuous or possibly associated with mild hemolysis.166,167,168
Ribosephosphate isomerase catalyzes the interconversion of ribulose-5-phosphate and ribose-5-phosphate. Deficiency of the enzyme has been described as one of the rarest human disorders.169 It manifests with progressive leukoencephalopathy and neuropathy. No dysfunction of red cells was reported.170
Ribulose-phosphate epimerase converts ribulose-5-phosphate to xylulose-5-phosphate. The exact activity of this enzyme in human hemolysates has not been reported but seems to be less than that of ribosephosphate isomerase.
Transketolase effects the transfer of two carbon atoms from xylulose-5-phosphate to ribose-5-phosphate, resulting in the formation of the 7-carbon sugar sedoheptulose-7-phosphate and the 3-carbon sugar glyceraldehyde-3-phosphate.171,172,173 It can also catalyze the reaction between xylulose-5-phosphate and erythrose-4-phosphate, producing fructose-6-phosphate and glyceraldehyde-3-phosphate. Thiamine pyrophosphate is a coenzyme for transketolase, and the activity of erythrocyte transketolase is used as an index of the adequacy of thiamine nutrition.174
The conversion of seduhepulose-7-phosphate and glyceraldehyde-3-phosphate into erythrose-4-phosphate and fructose-6-phosphate is catalyzed by transaldolase.175 This is another one in the series of molecular rearrangements that leads in the conversion of the 5-carbon sugar formed in the phosphogluconate dehydrogenase step to metabolic intermediates of the EMP. Transaldolase deficiency was first reported in 2001 as a new inborn error of the pentose phosphate pathway.176 To date, 23 patients from 13 families have been described.177,178 It is a pleiotropic metabolic disorder, and patients present in the neonatal or antenatal period with dysmorphic features, hepatosplenomegaly, abnormal liver function, cardiac defects, thrombocytopenia, bleeding tendencies, and anemia. The latter appears to be hemolytic in nature, possibly because of decreased levels of NADPH.179,180
Red cells contain L-hexonate dehydrogenase, an enzyme that has the capacity to reduce aldoses such as glucose, galactose, or glyceraldehyde to their corresponding polyol (i.e., glucose to sorbitol, galactose to dulcitol, and glyceraldehyde to glycerol). NADPH serves as a hydrogen donor for this reaction.181 Aldose reductase is another enzyme that can catalyze this reaction. It is present in red cells,182 and increased levels have been implicated in diabetic complications, such as retinopathy183 and autonomic neuropathy.184
The red cell has the capacity to use several other substrates in addition to glucose as a source of energy. Among these are adenosine, inosine, fructose, mannose, galactose, dihydroxyacetone, and lactate.185 Although in the circulation red cells normally rely on glucose as their energy source, the use of other substrates, particularly during blood storage (Chap. 138) and in certain experimental situations, is of interest.
The red cell contains a high concentration of the sulfhydryl-containing tripeptide GSH. It serves a major role in antioxidant defense, detoxification, and maintenance of thiol status. Reported concentrations range between 0.4 and 3.0 mM, with a terminal half-life (T1/2) of approximately 4 days.187 The wide interindividual range suggests that GSH levels are, at least in part, genetically determined.188 In its role of defense against oxidative stress GSH is oxidized to glutathione disulfide (GSSG), which can be reverted back to GSH by mediation of glutathione reductase. In addition GSSG can be transported out of the cell.
Glutathione biosynthesis occurs in two ATP-dependent steps:
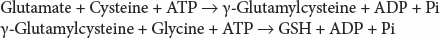
The first step is rate limiting and catalyzed by glutamate cysteine ligase (GCL) (γ-glutamylcysteine synthetase). Feedback inhibition of GCL by GSH is commonly considered a key regulatory step in GSH homeostasis, but other pathways are likely to play a role in maintaining GSH levels.189 GCL is a heterodimer composed of a catalytic 73-kDa catalytic subunit (GCLC) and a 31-kDa modifier subunit (GCLM).190,191 An intersubunit disulfide bond has been implicated in stabilization and catalytic efficiency of the heterodimer, acting as a cellular redox switch to couple enzyme activity, and consequently GSH levels, with the reduction-oxidation (redox) state of the cell.192,193 An alternative model suggests that increased levels of oxidative stress induce the formation of high activity heterodimer complexes from low activity monomeric and holoenzyme forms of the enzyme.194 GCL subunits are encoded by separate genes, located on chromosome 6p12 (GCLC) and 1p22.1 (GCLM), respectively. GCLC contains 16 exons, encoding for the 637-amino-acids catalytic subunit, whereas GCLM contains 7 exons that encode the 274 amino acids of the modifier subunit. A trinucleotide repeat polymorphism in the 5′-UTR affects GCLC gene expression and is associated with alterations in GCL activity and GSH levels.195 GCL deficiency is a very rare cause of hemolytic anemia.
The second step in GSH synthesis is irreversible and mediated by glutathione synthetase (GS). This enzyme is a homodimer composed of 52-kDa subunits,196 and the crystal structure of human GS has been resolved.197 There is one gene coding for GS. This 23-kb gene (GSS) is located on chromosome 20q11.2 and contains 13 exons, of which the first is noncoding, that code for the 474-amino-acids-long protein. Deficiency of GS is the most common disorder of GSH synthesis and is associated with hemolytic anemia.
One important function of GSH in the erythrocyte is the detoxification of low levels of hydrogen peroxide that may form spontaneously or as a result of drug administration. Hydrogen peroxide is reduced to water through mediation of the enzyme glutathione peroxidase,198 thereby oxidizing GSH to GSSG (see Fig. 47–7). Several glutathione peroxidases exist but only type 1 appears to be expressed in red blood cells.199 Glutathione peroxidase is a selenium-containing200 tetrameric enzyme consisting of 21-kDa subunits. A polymorphism affecting the activity of this enzyme, which is most common in persons of Mediterranean descent,201 has been described. The consequent decreases in enzyme activity are without clinical effect.202 In agreement with this, complete loss of glutathione peroxidase activity in mice was found to be without any consequences, even at high levels of oxidative stress, thus suggesting that glutathione peroxidase is of minor significance for red cell function.199
GSH also functions in maintaining integrity of the erythrocyte by reducing sulfhydryl groups of membrane proteins,203 and glycolytic enzymes,204 for example, PK.205 In the process of reducing peroxides or oxidized protein sulfhydryl groups, GSH is converted to GSSG, or may form mixed disulfides. Thus, for instance, GSSG has the capacity to inhibit red cell HK,49,206 although greater than physiologic levels may be needed for this effect. It may also complex with hemoglobin A to form hemoglobin A3.207
Glutathione reductase (GR) provides an efficient mechanism for the reduction of GSSG to regenerate GSH, and thus maintaining high intracellular levels of GSH. A mitochondrial and a cytoplasmic isozyme are both produced from the same mRNA, most likely by alternative initiation of translation.208 GR is a homodimer, linked by a disulfide bridge. Each 56-kDa subunit contains four domains, of which domains 1 and 2 bind FAD (flavin adenine dinucleotide) and NADPH, respectively, with domain 4 constituting the interface.209 The protein is encoded by the GR gene, located on chromosome 8p21.1. GR spans 50 kb, contains 13 exons, and encodes a 522-amino-acids-long protein. GR is a flavin enzyme, and either NADPH or NADH may serve as a hydrogen donor.210 In the intact cell, only the NADPH system appears to function.211 The same enzyme system appears to have the capacity to reduce mixed disulfides of GSH and proteins.42 The activity of red cell GR is strongly influenced by the riboflavin content of the diet,212 and its activity is used as a biomarker for riboflavin status.213 Correction of partial GR deficiency by riboflavin administration has been reported to ameliorate hemolysis in a case of unstable hemoglobin.214 Hereditary deficiency of GR is a very rare disorder and associated with hemolytic anemia.
Red cells also contain thioltransferase that can catalyze GSH-dependent reduction of some disulfides.215,216
Oxidized glutathione is actively extruded from the erythrocyte.217,218 GSSG efflux is likely an important regulator of GSH turnover in red blood cells.219 The system consists of at least two GSSG-activated adenosine triphosphatases (ATPases) that serve as an enzymatic basis for this transport process.220 In addition to transporting GSSG, the system appears to have the capacity to transport thioether conjugates of GSH and electrophiles formed by the action of glutathione-S-transferase.221,222 Preliminary evidence indicates that multidrug resistance protein 1 (MRP1) may be the exporter protein of both glutathione-S conjugates and GSSG.189,223 Erythrocytes contain glutathione-S-transferases rho, sigma, and theta.224,225,226 Glutathione-S-transferase catalyzes the formation of a thioether bond between GSH and a variety of xenobiotics. The role of glutathione-S-transferase in the erythrocyte has not been established. It may be that it serves to cleanse the blood of xenobiotics to which the red cell membrane is permeable. Glutathione-S-transferase could conjugate such substances to glutathione, and the detoxified product of conjugation would be transported out of the red cell for subsequent disposal. The enzyme has the capacity to reversibly bind heme, and a possible role in heme transport has been postulated.227 Deficiency of this enzyme is associated with hemolytic anemia, but a cause-and-effect relationship has not been established.228
Superoxide radicals that are formed are converted to hydrogen peroxide by the action of the copper-containing enzyme superoxide dismutase (SOD).229 Red blood cells contain SOD type 1 (SOD1). Mutations in SOD1 are associated with the dominant disorder familial amyotrophic lateral sclerosis.230 Patients display no hematologic phenotype. In agreement with this, studies on SOD1-deficient mice have shown that a 50 percent reduced SOD activity in red cells is probably sufficient to exert its protective effects.231 SOD1 null mice are viable, but show elevated levels of oxidative stress that causes regenerative anemia and triggers autoantibody production.232
Hydrogen peroxide can be also decomposed to water and oxygen by catalase and peroxiredoxin. Both enzymes are abundantly present in the red blood cell, suggesting an important role in the cell’s oxidative defense. Nevertheless, acatalasemia, or catalase deficiency, is a rare benign condition without hematologic consequences. It has been suggested, though, that it may act as a complicating factor in age and diseases such as diabetes233 and oxidative stress-related conditions.234 Deficiencies of peroxiredoxin 2, the major red cell peroxiredoxin,235 have not been reported in humans. Peroxiredoxin 2 null mice, however, develop severe hemolytic anemia with Heinz body formation,236 and show signs of abnormal erythropoiesis.237 Based on mouse models, it has been postulated that glutathione peroxidase, catalase, and peroxiredoxin each have distinct roles in the scavenging of hydrogen peroxide and antioxidative defense.238,239,240
Approximately 97 percent of the total nucleotide content of the mature red blood cell consists of interconvertible adenosine phosphates (Chap. 31). Less than 3 percent of total nucleotides are guanosine phosphates. ATP is the most abundant adenosine phosphate (comprising roughly 84 percent of total adenosine ribonucleotides), whereas ADP (14 percent) and adenosine monophosphate (AMP, 1 percent) are present in considerably lower amounts. The interconversion of adenine nucleotides is modulated by adenylate kinase (AK):
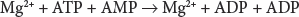
By catalyzing the reversible phosphoryl transfer among ATP, ADP, and AMP, AK contributes to cellular adenine nucleotide homeostasis. Red cells contain the AK1 isozyme, which is present in the cytosol as a monomeric enzyme composed of 194 amino acids. The recombinant purified enzyme has a molecular mass of approximately 22 kDa.241 The AK1 gene is localized on chromosome 9q34.1, and consists of 7 exons of which exon 1 is noncoding. AK activity depends on the presence of Mg2.
ATP serves as a cofactor in a number of reactions, such as the phosphorylation steps mediated by HK and PFK in glycolysis, the synthesis of GSH, and ATPase-dependent function of membrane pumps. Therefore, ATP is crucial in maintenance of the red cell’s structure and function. Because the mature red cell is unable to synthesize adenosine phosphates from precursor molecules, it relies on salvage pathways to preserve adenosine ribonucleotides. This is of particular importance for AMP because this adenosine ribonucleotide is at risk of being lost from the adenine pool by dephosphorylation to adenosine and, subsequent, irreversible deamination to inosine by the enzyme adenosine deaminase (ADA). ADA thus plays a regulatory role in the concentration of adenosine ribonucleotides in the red cell. The gene encoding ADA (ADA) is located on chromosome 20q13.12. It comprises 12 exons that encode a 363-amino-acid protein.
Deficiency of AK and hyperactivity of ADA are both very rare causes of hereditary nonspherocytic hemolytic anemia.
Additional enzymes of purine metabolism are also present in the red cell. Although disorders of these enzymes are associated with a number of metabolic diseases, their function does not appear to be relevant for the red blood cell as these disorders are without hematologic consequences.242
Pyrimidine ribonucleotides are found only in trace amounts in the mature red blood cell. They are lost from the cell together with the degradation of ribosomes and RNA during reticulocyte maturation. Pyrimidine-5′-nucleotidase-1 (P5′N1) mediates this loss by catalyzing the dephosphorylation of pyrimidine nucleoside monophosphates into the corresponding nucleosides (cytidine and uridine), which are freely diffusible across the membrane.243 P5′N1 is specific for pyrimidine nucleotides and does not use purine nucleotides as substrate.244 The enzyme requires Mg2+ for it activity, and is inhibited by a number of heavy metals, including Pb2+.245 Like other red blood cell enzymes, P5′N1 activity is much higher in reticulocytes. The enzyme declines in activity during red cell aging.246 P5′N1 also has phosphotransferase properties, suggesting an additional role of this enzyme in nucleotide metabolism.247 The crystal structure of mouse P5′N1 has been published, providing a framework for understanding the kinetics of both nucleotidase and phosphotransferase activities of human P5′N1.248
P5′N1 is encoded by the NT5C3A gene on chromosome 7p14.3. It comprises 11 exons, and produces three distinct mRNAs by alternative splicing. Red cell P5′N1 is translated from the mRNA lacking exons 2 and R.249,250 It is a 286-amino-acids-long monomeric protein with an apparent molecular weight of 34 kDa.249
A second P5′N is present in red blood cells, the activity of which is generally measured together with that of P5′N1. This enzyme (P5′N2) is encoded by a separate gene, shows little homology to P5′N1, and is not strictly pyrimidine-specific. It is unable to compensate for deficient function of P5′N1.247,251 P5′N1 deficiency is one of the most common causes of hereditary nonspherocytic hemolytic anemia.
Human red blood cells have been found to express low NAD synthesis activity, mediated by nicotinamide mononucleotide adenylyltransferase.252 It appears that the predominant isozyme in red blood cells is nicotinamide mononucleotide adenylyltransferase-3.253 Human dysfunction of this enzyme has not been reported but the deficiency in mice blocks glycolysis at the GAPDH step and is associated with hemolytic anemia.254
GENETICS
The great majority of red cell enzyme deficiencies that cause hemolytic anemia are hereditary. Most are inherited as autosomal recessive disorders, but G6PD deficiency and PGK deficiency are X-chromosome-linked. The vast majority of the genes encoding for the red cell enzymes have been identified, making the molecular diagnosis of hereditary red cell enzyme deficiency possible. Occasionally, acquired forms of enzyme deficiencies, particularly PK deficiency, have been encountered, usually in patients with hematologic neoplasia.255,256,257,258,259
Table 47–2 lists the erythrocyte enzyme deficiencies shown to cause hemolytic disease. Other red cell enzyme deficiencies (Table 47–3) do not appear to cause hemolysis or other functional abnormality of the erythrocyte.202 For example, acatalasemia, the state in which there is a virtually total absence of red cell catalase, is devoid of hematologic manifestations.233 Similarly, red cells without acetylcholinesterase survive normally in most cases.260
| Diagnosis (Reference) | |||||||
|---|---|---|---|---|---|---|---|
| Enzyme | Clinical Features | Inheritance | Red Cell Morphology | Screening Test | Assay | Response to Splenectomy* | Approximate Frequency† |
| Hexokinase | HNSHA | AR | Unremarkable | — | 580 | + + | Rare |
| Glucose phosphate isomerase | HNSHA; neurologic abnormalities | AR | Unremarkable | 580 | 580 | + + + | Unusual |
| Phosphofructokinase | HNSHA and/or muscle glycogen storage disease | AR | Unremarkable | — | 580 | 0 | Rare |
| Aldolase | HNSHA and mild liver glycogen storage; myopathy, mental retardation | AR | Unremarkable | — | 580 | ? | Very rare |
| Triosephosphate isomerase | HNSHA and severe neuromuscular disease | AR | Unremarkable | 580 | 580 | ? | Rare |
| Phosphoglycerate kinase | HNSHA; myoglobinuria neuromuscular disorder | SL | Unremarkable | — | 580 | + + | Rare |
| Pyruvate kinase | HNSHA | AR | Usually unremarkable; occasionally contracted echinocytes | 580 | 580 | + + | Unusual |
| Glucose-6-phosphate dehydrogenase | HNSHA; drug- or infection-induced hemolysis; favism | SL | Usually unremarkable; rarely “bite cells” | 580 | 580 | ± | Very common |
| Glutathione reductase | Drug-sensitive hemolytic anemia and favism | AR | Unremarkable | 580 | 580 | ? | Very rare |
| Glutamate cysteine synthetase | HNSHA; drug- or infection-induced hemolysis; neurologic abnormalities | AR | Unremarkable | 644 | 645 | ? | Very rare |
| Glutathione synthetase | HNSHA; drug- or infection-induced hemolysis; neurologic defect and 5-oxoprolinuria in some cases | AR | Usually unremarkable | 644 | 645 | 0 | Rare |
| Pyrimidine 5′-nucleotidase | HNSHA; mental retardation in some cases | AR | Prominent stippling | 646 | 647 | 0 | Unusual |
| Adenylate kinase | HNSHA | AR | Unremarkable | — | 580 | ? | Rare |
| Adenosine deaminase (increased activity) | HNSHA | AD | Unremarkable | — | 580 | ? | Very rare |
| Enzyme | Clinical Features | Inheritance | Diagnosis Reference Assay | Estimated Frequency* | Reference |
|---|---|---|---|---|---|
| 6-Phosphogluconate dehydrogenase (complete deficiency) | None | AR | 580 | Unusual | 166,167,168 |
| 6-Phosphogluconolactonase (partial defect) | Probably none | AD | 648 | Unusual | 163 |
| δ-ALA dehydrase | None | AD | 649 | ||
| Acetylcholinesterase | None | AR | 580 | Very rare | 260 |
| Adenine phosphoribosyl transferase | Kidney stones | AR | 650 | Rare | 651 |
| Adenosine deaminase (decreased activity) | Immunodeficiency | AR | 580 | Rare | 652 |
| AMP deaminase | None | AR | 653 | Unusual | 654 |
| Bisphosphoglycerate mutase | Erythrocytosis | AR | 580 | Very rare | 377,380 |
| Carbonic anhydrase I | None | AR | 655 | Rare | 656 |
| Carbonic anhydrase II | Osteoporosis | AR | Rare | 657 | |
| Catalase | Oral ulcers in some types | AR | 580 | Rare | 658 |
| Cytochrome-b5-reductase | Methemoglobinemia; mental retardation | AR | 580 | Unusual | 659 |
| Enolase | HNSHA? | AD? | 580 | Rare | 115,116 |
| Galactokinase | Cataracts | AR | 580 | Rare | 660 |
| Galactose-1-P-uridyltransferase | Cataracts; mental retardation; liver disease | AR | 580 | Rare | 661 |
| Glutathione peroxidase (partial deficiency) | None | AR and AD662 | 580 | Very common | 580,662 |
| Glutathione reductase (partial deficiency) | None | Usually not inherited662 | 580 | Very common | 662,663 |
| Glutathione-S-transferase | HNSHA | ? | 580 | Very rare | 228 |
| Glyceraldehyde-3-phosphate dehydrogenase (partial defect) | None | AD | 580 | Unusual | 94 |
| Glyoxalase I | None | AR | Rare | 664 | |
| Hypoxanthine-guanine phosphoribosyl transferase (HGPRT) | Lesch-Nyhan syndrome (neurologic symptoms and gout) | SL | 665 | Rare | 666 |
| Inosine triphosphatase | None | AR | 656 | Rare | 667 |
| Lactate dehydrogenase | None | AR | 580 | Rare | 135 |
| NADPH diaphorase | None | AR | 580 | Rare | 668 |
| Phosphoglucomutase | None | AR | 580 | Rare | 669 |
| Transaldolase | Liver disease, thrombocytopenia, HNSHA? | AR | 670 | Rare | 175 |
| Uroporphyrinogen 1 synthase | Porphyria | AD | 671 | Unusual (common in selected populations) | 672 |
The lack of clinical manifestations is not always clearcut. In some instances, hemolytic anemia is reported in some individuals with a given deficiency but not in others. For example, most subjects with LDH deficiency have no anemia, but cases with hemolysis have been reported.136 Such ambiguity could result from differences in environmental and genetic factors or from bias of ascertainment. Erythrocyte enzyme assays are usually performed on patients with hemolytic anemia. Thus, a benign enzyme defect may be thought, mistakenly, to cause hemolysis because it is found in a patient with hemolytic anemia caused by an unrelated and undetected defect. Deficiencies of PGK, GS, or AK are usually associated with hereditary nonspherocytic hemolytic anemia, but cases have been reported in which these deficiencies were unassociated with any hematologic manifestations.261,262,263 At times it has been suggested that moderate decreases in the activity of glutathione peroxidase causes hemolytic anemia, but the best available evidence indicates that this enzyme is not ordinarily rate limiting in erythrocyte metabolism and not associated with hemolytic anemia.264 Table 47–3 includes deficiencies that may cause hemolytic anemia but for which a cause-and-effect relationship has not been clearly established, such as those of phosphogluconolactonase,163 enolase,115,116 and glutathione-S-transferase.228
Patients with unstable hemoglobins (Chap. 49) may present with the clinical picture of hereditary nonspherocytic hemolytic anemia. Hemolytic anemia resulting from abnormalities in the lipid composition of the red cell membrane, particularly increased phosphatidyl choline, occurs rarely (Chap. 46).
The “normal” or wild-type enzyme is designated as G6PD B. Many variants of G6PD have been detected all over the world, associated with a wide range of biochemical characteristics and phenotypes. Accordingly, five classes of G6PD variants can be distinguished based on enzymatic activity and clinical manifestations (Table 47–4).265 Before it became possible to characterize G6PD variants at the DNA level, they were distinguished from each other on the basis of biochemical characteristics, such as electrophoretic mobility, Km for NADP and glucose-6-P, ability to use substrate analogues, pH activity profile, and thermal stability. To facilitate comparison of variants characterized in different laboratories, international standards for the methodology were established.266 In the case of the common G6PD A– and G6PD Mediterranean mutations, the abnormal enzyme may be synthesized at normal or near-normal rates but has decreased stability in vivo.267 The amount of enzyme antigen in the red cells declines concurrently with enzyme activity.268 This suggests that the mutant protein in these variants is rendered unusually sensitive to proteolysis in the environment of the erythrocyte.269 Other mutations also result in the formation of enzyme molecules with decreased enzyme activity268 and with altered kinetic properties,270 some of which may render them functionally inadequate.158 By far the majority of mutations (85 percent) are missense mutations causing the substitution of a single amino acid.158 More severe mutations such as frameshift and nonsense mutations have not been found, indicating that some residual activity is required for survival. In agreement with this, targeted deletion of G6PD in the mouse causes embryonic lethality.271
| Variant | Nucleotide Substitution | Amino Acid Substitution | WHO Class† | Distribution | Reference |
|---|---|---|---|---|---|
| Gaohe | c.95A>G | p.(His32Arg) | III | Chinese | 673 |
| Honiara | c.99A>G c.1360C>T | p.(Ile33Met) p.(Arg454Cys) | I | Solomon Islands | 674 |
| Orissa | c.131C>G | p.(Ala44Gly) | III | India, Italy | 675,676 |
| Aures | c.143T>C | p.(Ile48Thr) | III | Algeria, Tunisia | 677,678 |
| Metaponto | c.172G>A | p.(Asp58Asn) | III | Italy | 679 |
| A– | c.202G>A c.376A>G | p.(Val68Met) p.(Asn126Asp) | III | Africa | 277 |
| Namoru | c.208T>C | p.(Tyr70His) | II | Vanuatu Archipelago | 680 |
| Ube-Konan | c.241C>T | p.(Arg81Cys) | III | Japan, Italy | 676,681 |
| A+ | c.376A>G | p.(Asn126Asp) | III-IV | Africa, Mediterranean | 272 |
| Vanua Lava | c.383T>C | p.(Leu128Pro) | II | Southwestern Pacific | 680 |
| Quing Yan | c.392G>T | p.(Gly131Val) | III | China | 682 |
| Mahidol | c.487G>A | p.(Gly163Ser) | III | Southeast Asia | 683 |
| Santamaria | c.542A>T c.376A>G | p.(Asp181Val) p.(Asn126Asp) | II | Costa Rica, Italy | 684,685 |
| Mediterranean, Dallas, Panama, Sassari | c.563C>T | p.(Ser188Phe) | II | Mediterranean | 277,686 |
| Coimbra | c.592C>T | p.(Arg198Cys) | II | India, Portugal | 687 |
| A– | c.680G>T c.376A>G | p.(Arg227Leu) p.(Asn126Asp) | III | Africa | 274 |
| Seattle, Lodi, Modena, Ferrara II, Athens-like | p.(Asp282His) | III | United States, Italy | 688,689,690 | |
| Montalbano | c.854G>A | p.(Arg285His) | III | Italy | 691 |
| Viangchan, Jammu | c.871G>A | p.(Val291Met) | II | China | 692,693 |
| Kalyan, Kerala, Jamnaga, Rohini | c.949G>A | p.(Glu317Lys) | III | India | 694,695 |
| A–, Betica, Selma, Guantanamo | c.968T>C c.376A>G | p.(Leu323Pro) p.(Asn126Asp) | III | Africa, Spain | 274 |
| Chatham | c.1003G>A | p.(Ala335Thr) | II | Italy, Asia, Africa | 277 |
| Chinese-5 | c.1024C>T | p.(Leu342Phe) | III | China | 682 |
| Ierapetra | c.1057C>T | p.(Pro353Ser) | II | Greece | 696 |
| Cassano | c.1347G>C | p.(Gln449His) | II | Italy, Greece | 697,698 |
| Union, Maewo, Chinese-2, Kalo | c.1360C>T | p.(Arg454Cys) | II | Italy, Spain, China, Japan | 697,699,700 |
| Canton, Taiwan-Hakka, Gifu-like, Agrigento-like | c.1376G>T | p.(Arg459Leu) | II | Japan, Italy | 701,702 |
| Cosenza | c.1376G>C | p.(Arg459Pro) | II | Italy | 697 |
| Kaiping, Anant, Dhon, Sapporo-like, Wosera | c.1388G>A | p.(Arg463His) | II | China | 700,702 |
Detailed biochemical and genetic characteristics of some 400 putatively distinct G6PD variants and more than 200 different mutations have been tabulated.155,160 Table 47–4 lists common G6PD variants that have reached polymorphic frequencies in certain populations.
Among persons of African descent, a mutant enzyme G6PD A+, with normal activity is polymorphic. It migrates electrophoretically more rapidly than the normal B enzyme, has substitution of Asn to Asp at codon 126, resulting from nucleotide change c.376A>G.272 G6PD A– is the principal deficient variant found among people of African origin. The red cells contain only 5 to 15 percent of the normal amount of enzyme activity; however because of the instability of the enzyme, the age-dependent decline of the activity renders old red cells severely deficient and susceptible to hemolysis. These two electrophoretically rapid variants are common in African populations have in common a nucleotide substitution at cDNA nucleotide 376 that produces the amino acid substitution responsible for the rapid electrophoretic mobility. Most samples with G6PD A– manifest an additional in cis G>A mutation at cDNA nucleotide 202 (c.202G>A; p.Val68Met), which accounts for its in vivo instability.273 Less commonly, the additional mutation is at a different site (c.680G>T or c.968T>C).273 Thus G6PD A– arose in an individual who already had the G6PD A+ mutation. However, the ancestral human sequence has been deduced to be that of G6PD B, both by showing that this is the sequence of the chimpanzee,274 our nearest relative, and by analysis of linkage dysequilibrium.275 Although it has been suggested that only the interaction of p.Val68Met and p.Asn126Asp invariably results in G6PD A– deficiency,276 the c.202G>A mutation has been found in a patient to cause deficiency without the presence of the mutation at cDNA nucleotide (nt) 376.274
Among white populations, G6PD deficiency is most common in Mediterranean countries. The most common enzyme variant in this region is G6PD Mediterranean.270,277 The enzyme activity of the red cells of individuals who have inherited this abnormal gene is barely detectable. Other variants are also prevalent in the Mediterranean region, including G6PD A– and G6PD Seattle (see Table 47–4).
A great many different variants have been described in Asian populations. Some of these proved to be identical at a molecular level (e.g., G6PD Gifu-like, Canton, Agrigento-like, and Taiwan-Hakka all have the same mutation at cDNA nt 1376 [see Table 47–4]). DNA analysis shows that more than 100 different mutations are found in various Asian populations.160,278
Some mutations of G6PD result in chronic hemolysis without, but exacerbated by, precipitating causes. These variants are class I mutants (World Health Organization [WHO] class 1).265 From a functional point of view, these mutations are more severe than the more commonly occurring polymorphic forms of the enzyme, such as G6PD Mediterranean and G6PD A–. On a molecular level, such variants are often caused by mutations located in exons 10 and 11, encoding the subunit interface, or affect residues that bind the structural NADP molecule.143,158 There are, however, exceptions to this rule.28,279,280,281 The clinical severity of these variants can be quite variable.282
G6PD deficiency has also been encountered in the rat, dog,283 mouse,284 and horse.285 G6PD deficiency in mice has been rescued by stable in vivo expression of the human G6PD gene in hematopoietic tissues by a gene transfer approach.271,286
PK deficiency is the second most common enzyme disorder in glycolysis and the most common cause of nonspherocytic hemolytic anemia.287 Like G6PD deficiency, the disease is genetically heterogeneous, with different mutations causing different kinetic changes in the enzyme that is formed. There are even cases in which the activity of PK as measured in vitro is higher than normal, but a kinetically abnormal enzyme is responsible for the occurrence of hemolytic anemia.288 Kinetic characterization and analysis of PK mutants is considerably more complex than analysis of G6PD mutants. Most PK-deficient patients are compound heterozygous for two different (missense) mutations, rather than homozygous for one. Assuming that stable mutant monomers are synthesized, up to seven different tetrameric forms of PK may be present in compound heterozygous individuals, each with distinct structural and kinetic properties. This complicates genotype-to-phenotype correlations in these individuals as it is difficult to infer which mutation is primarily responsible for deficient enzyme function and the clinical phenotype.289,290 More than 230 mutations in the PKLR gene encoding the red cell PK have been identified. Seventy percent of these mutations are missense mutations affecting conserved residues in structurally and functionally important domains of PK. There appears to be no direct relationship between the nature and location of the substituted amino acid and the type of molecular perturbation.124 Hence, the nature of the mutation has relatively little predictive value with respect to the severity of the clinical course and the phenotypic expression of identical mutations can be strikingly different in patients.29,289,290,291
Apart from decreased red blood cell survival ineffective erythropoiesis because of increased numbers of apoptotic cells is implicated as one of the pathophysiologic features of PK deficiency.292,293 In particular, glycolytic inhibition by mutation of PKLR has been suggested to augment oxidative stress, leading to proapoptotic gene expression.293
PK deficiency may be also caused by mutations not directly involving the PKLR gene as demonstrated by a deficiency of PK being one of the key features of severe congenital hemolytic anemia caused by mutations in the key erythroid transcription factor KLF1.294
There is evidence that PK deficiency provides protection against infection and replication of Plasmodium falciparum in human erythrocytes,295,296 an effect possibly mediated by reduced ATP levels in PK-deficient red blood cells.297 This suggests that PK deficiency may confer a protective advantage against malaria in human populations in areas where this disease is endemic. In agreement with this, population studies on sub-Saharan African populations indicate that malaria is acting as a selective force in the PKLR genomic region.298,299,300
PK deficiency has also been recognized in mice, dogs, and multiple breeds of domestic cats.301 In all these animals, the deficiency causes severe anemia and marked reticulocytosis, closely resembling human PK deficiency. Basenji dogs with PK deficiency completely lack PKLR enzymatic activity and, instead, only the PK-M2 isozyme is expressed in their red blood cells.302 A unique feature of PK deficiency in dogs is the progressive development of myelofibrosis and osteosclerosis. Marrow fibrosis may occur in response to damage caused by iron overload,303 although factors associated with marked erythropoiesis have also been proposed to play a role.304 Gene therapy approaches have been employed to cure PK deficiency in dogs.305 PK-deficient mice show delayed switching from PK-M2 to PK-R, resulting in delayed onset of the hemolytic anemia.306 PK deficiency in mice has been rescued by expression of the human PK-R isozyme in murine hematopoietic stem cells.307,308
Nineteen families with HK deficiency have been described as of the time of this writing309,310,311 and only four patients have been characterized at the molecular level.310,311,312,313 Two of these patients were homozygous, either for a highly conserved substitution in the enzyme’s active site313 or a lethal out-of-frame deletion of exons 5 to 8 of HK1.310 In one patient a regulatory mutation was identified in the putative erythroid-specific promoter. In vitro, this mutation disrupted binding of the AP-1 transcription factor complex, leading to strongly decreased gene expression.311
In mice, a mutation designated downeast anemia causes severe hemolytic anemia with extensive tissue iron deposition and marked reticulocytosis, representing a mouse model of generalized HK deficiency.314
Glucosephosphate isomerase deficiency is second to PK deficiency in frequency, with respect to glycolytic enzymopathies. To date, approximately 55 families with glucosephosphate isomerase deficiency have been described worldwide.315,316,317,318,319,320 Most of these patients are compound heterozygous for mutations that partially inactivate the enzyme. Most of the 31 GPI mutations reported to date are missense mutations. Mapping of these mutations to the crystal structure of the human enzyme and recombinant expression of genetic variants has provided considerable insight in the molecular mechanisms causing hemolytic anemia in this disorder.321,322 The majority of the mutations disrupt key interactions that contribute directly or indirectly to the architecture of the enzyme’s active site.321 In rare cases, GPI deficiency also affects nonerythroid tissues, causing severe neuromuscular symptoms and granulocyte dysfunction.323,324,325,326,327,328 The finding that GPI also functions as a neuroleukin,329 an autocrine motility factor,330 a nerve growth factor,331 and a differentiation and maturation mediator332 has led to the hypothesis that the mutation-dependent loss of cytokine function of GPI could account for the neuromuscular symptoms.333 An alternative explanation involves disturbed glycerolipid biosynthesis in GPI deficiency, which could have significant effects on membrane formation, membrane function, and axonal migration.334,335
Homozygous GPI-deficient mice exhibit hematologic features resembling that of the human enzymopathies. In addition, other tissues are also affected, indicating a reduced glycolytic capability of the whole organism.336 Complete loss of GPI in mice is embryonically lethal.337
Because red cells contain both PFK M and L subunits, mutations affecting either gene (PFKM or PFKL) will lead to a partially reduced red cell enzyme activity in PFK deficiency. Mutations in the PFKM
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


