INTRODUCTION
SUMMARY
Epigenetics involves a heritable change in phenotype without a change in genotype–with the inheritance of particular chromatin and transcription states often underlying the mechanism. Chromatin regulates gene expression by controlling the density and positioning of nucleosomes, and by the use of histone- and DNA-modifying enzymes. Chromatin and transcription factors drive proper differentiation decisions through their coregulation of key factors in development and proliferation. Of particular interest to hematologists are instances when misregulation/mutation of chromatin factors drives hematologic malignancies and myeloproliferative disorders. Here, fusion proteins that involve the mistargeting of chromatin regulators have been known for decades. More recently, high-throughput sequencing and other genomics approaches have revealed mutations in many types of chromatin regulators in hematologic malignancies, including mutations in chromatin remodelers, DNA methylation regulators, histone modification enzymes, and metabolic enzymes affecting epigenetic cofactors. Overall, these studies reveal a consistent theme: epigenetic and genetic mutations confer both variation and plasticity to the transcriptome, and when combined with selection, arrive at transcriptomes that promote proliferation, survival, and adaptability. This chapter addresses these mechanistic principles of chromatin, and their misregulation in hematologic malignancies, as well as emerging therapeutic approaches.
Acronyms and Abbreviations:
AF, ALL1-fused gene; ALL, acute lymphocytic leukemia; AML, acute myeloid leukemia; BAF, BRG/BAF-associated factors; BCL, B-cell lymphoma family of regulator proteins that regulate cell death; BET, bromo and extraterminal; CHD, chromodomain remodeler; CMML, chronic myelomonocytic leukemia; DNAme, DNA methylation; DNMT, DNA methyltransferase; DOT1, a histone H3 methyltransferase; EGR1, early growth response protein 1; EZH2, enhancer of zeste homologue 2; H3, histone H3; HAT, histone acetyltransferase; HDAC, histone deacetylase; HIF, hypoxia-inducible transcription factor; 5hmC, 5-hydroxymethylcytosine; HMT, histone methyltransferase; HSC, hematopoietic stem cell; IDH, isocitrate dehydrogenase; Ifng promoter, interferon-γ promoter; ISWI, imitation SWI remodeler; MBD, methyl-domain binding; 5mC, 5-methylcytosine; MLL, mixed lineage leukemia; MTA, metastasis-associated; NuRD, nucleosome remodeling and deacetylation factor; NURF, nucleosome remodeling factor; 2OG, 2-oxoglutarate; PRC2, polycomb repressive complex 2; R-2HG, (R)-2-hydroxyglutarate; RAR, retinoic acid receptor; RNAP II, RNA polymerase II; SDH, succinate dehydrogenase; SRF, serum response factor; SWI/SNF, switch and sucrose nonfermenting remodeler; TDG, thymine DNA glycosylase; UHRF1, ubiquitin-like with PHD and ring finger domains; UTX, X-chromosome encoded ubiquitously transcribed tetratricopeptide repeat.
DEFINITION AND OVERVIEW
Epigenetics is defined as a heritable change in phenotype without a change in genotype. Although epigenetic mechanisms vary, this chapter focuses on the most common mechanism: chromatin. Changes in chromatin/epigenetics accompany many steps in transcription, replication, and recombination. However, the aspects of highest interest and relevance involve examples where epigenetic factors and enzymes drive differentiation decisions, and where misregulation/mutation of these factors drives pathologies, such as hematologic malignancies. This decision making must be precise, as differentiation along the lymphoid and myeloid lineages involves the regulated generation of multiple cell types in temporal order and proper proportion. Decisions are arrived through collaboration among signaling systems, transcription factors, and chromatin regulators-which together regulate the key genes governing self-renewal, differentiation, and survival. This chapter focuses on chromatin factors with central roles in these processes: ATP-dependent remodelers, DNA methylation (DNAme)/demethylation enzymes, and histone modification enzymes. As a complete treatment is beyond the scope of this chapter, the focus here will be conceptual, with particular examples provided to create a framework for understanding the many instances where chromatin factors influence decision-making.
Beyond their roles in normal blood cell development, misregulation of chromatin factors is now known to be common in hematologic malignancies. Indeed, high-throughput whole-genome and/or exome sequencing of leukemias and lymphomas has revealed mutations in many types of chromatin regulators, including mutations in chromatin remodelers, DNA methyltransferases (DNMT), and histone modification enzymes, as well as revealing fusion proteins that involve chromatin regulators.1 In certain instances, modeling in the mouse supports these epigenetic mutations as the main drivers of the cancer phenotype. In other instances, epigenetic mutations cooperate with (and likely enable) additional genetic mutations, which cooperate to impact proliferation, survival, and plasticity, which can enable both cancer progression and therapy resistance. However, as many chromatin regulators are enzymes, they may be more targetable than mutations in DNA binding transcription factors, providing new therapeutic approaches.2 This chapter expands on these concepts, addressing the mechanistic basis of chromatin misregulation in hematologic malignancies, as well as emerging therapeutic approaches.
CHROMATIN REMODELING AND DNA ACCESS
Chromatin has a major impact on gene expression, mediated through interplay with transcription factors. Sequence-specific DNA-binding transcription factors are the most important factors in defining whether and when a gene is transcribed, and also define the locations and character of chromatin regions, as they target chromatin remodeling and modifying proteins. However, the initial chromatin landscape can control whether transcription factors have access to the DNA at a particular gene/region. Access to DNA is deterred by nucleosomes, the main repeating unit of chromatin structure, which can block the binding sites of transcription factors to chromatin.3 Likewise, DNAme can also block the binding of transcription factors, many of which will not bind DNA if the cytosine in their binding site is methylated (DNAme is discussed more extensively in the section “DNA Methylation and Demethylation”). Thus, the nucleosome and DNAme landscape together define the initial “open versus closed” chromatin state of a region with which the current repertoire of transcription factors within that cell type must contend. However, this landscape is dynamic, as signaling systems can modify transcription factors and chromatin components, altering their activity and the landscape both through their binding, and through their recruitment of nucleosome remodelers and chromatin modifiers.3,4,5
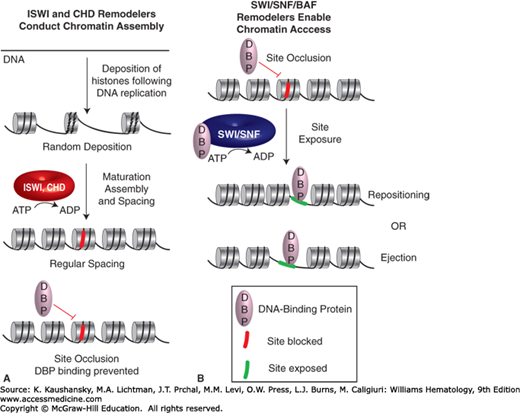
ATP-dependent chromatin remodeling complexes (termed hereafter remodelers) conduct central roles in regulating nucleosome occupancy and positioning (Fig. 12–1).3,4,5 For example, remodelers specialized for chromatin assembly (such as imitation SWI remodeler [ISWI]-family and chromodomain remodeler [CHD]-family remodelers) utilize ATP hydrolysis to facilitate tight-packed nucleosomes that lead to the occlusion of sites for site-specific DNA binding proteins, such as transcription factors. Access to chromatin at enhancers, promoters and other loci can be enabled by remodelers such as the switch and sucrose non-fermenting remodeler (SWI/SNF) complex, also termed the BRG/BAF-associated factors (BAF) complex, which can slide or eject the histone octamer, using the energy of ATP hydrolysis (Fig. 12–1). Notably, SWI/SNF can interact with (and facilitate the binding of) DNA-binding activators or repressors and can, therefore, help facilitate either activation or repression.3,4,5 Here, the ability of activators or repressors to interact with SWI/SNF complexes can be influenced by signaling cascades, which impart covalent modifications that enable or disable protein interactions. Taken together, ISWI and CHD remodelers often act to silencing genes via site blockage at enhancers and promoters, whereas SWI/SNF remodelers promote site exposure at those locations (Fig. 12–1), which is important for gene activation.
Figure 12–1.
Roles for ATP-dependent chromatin remodelers in chromatin assembly or chromatin access. Imitation SWI remodelers (ISWI)- and chromodomain remodelers (CHD)-family remodelers are involved in chromatin assembly genome-wide, and also interact with site-specific repressors to organize nucleosome spacing at genes, which can occlude sites for DNA-binding proteins. Switch and sucrose nonfermenting remodelers (SWI/SNF)-family remodelers conduct both nucleosome repositioning/sliding as well as ejection to expose DNA to DNA-binding proteins.
CHROMATIN REMODELING COMPLEXES IN BLOOD CELL DIFFERENTIATION
Clear roles for remodelers in blood cell differentiation are emerging. For example, SWI/SNF components affect the pool size of fetal hematopoietic stem cells (HSCs), and also impact HSC (and progenitor) proliferation and survival.6 SWI/SNF complex is also used for myeloid differentiation to granulocytes and for multiple steps in thymocyte development. More specifically, in mice SWI/SNF binds the interferon-γ (Ifng) promoter, and is required for its full transcription. Furthermore, mutations in the adenosine triphosphatase (ATPase) function of SWI/SNF are known to reduce β-globin expression and to prevent erythroid differentiation.7 Notably, B-cell lymphoma (BCL) factors BCL7A and BCL11B, which are considered members of SWI/SNF complex in many cell types, are common in hematologic malignancies; for example, mutations in BCL7A are found in approximately 20 percent of non-Hodgkin lymphoma and multiple myeloma cases, and mutations in BCL11B are found in 6 to 12 percent of T-cell acute lymphocytic leukemias (ALLs).8
Roles for ISWI- and CHD-family remodelers include roles for the well-characterized CHD-family remodeler nucleosome remodeling and deacetylation factor (NuRD), which interacts with histone deacetylase (HDAC) enzymes to silence genes. The metastasis-associated (MTA) subunits of NuRD help target NuRD subtypes to particular genes through their interaction with transcription factors and chromatin modifications.9 For example, in B-lymphocytes, MTA3 interacts with BCL6, a major regulator of B-cell differentiation, targeting NuRD repression and preventing terminal differentiation into plasma cells.10 Remarkably, expressing BCL6 in plasma cells while MTA3 is functional results in a reversion of the cell fate and reprogramming into B lymphocytes.10 Furthermore, the recruitment of the ISWI-family complex nucleosome remodeling factor (NURF) to the early growth response protein 1 (EGR1) locus (important for thymocyte maturation) involves interaction with the transcription factor serum response factor (SRF) by the NURF subunit BPTF, enabling its stable binding to promoters.11 Notably, Ikaros (which drives lymphoid differentiation) acts to inhibit both the ATP-dependent remodeling and HDAC activities of NuRD at target genes to enable activation rather than silencing.12 Taken together, these and other examples illustrate the use of remodeler function and recruitment to activate or repress key genes in blood differentiation.
PRINCIPLES OF HISTONE MODIFICATION
The process of transcriptional regulation is accompanied by the ordered placement of particular histone modifications at enhancers, promoters, and coding regions. There are dozens of different modifications that occur on histones, with the most common modifications being acetylation, methylation, ubiquitylation, and phosphorylation. An inventory and functional analysis of all of these modifications, the enzymes that place and remove these modifications, is beyond the scope of this chapter; however, more important are the concepts, which can then be applied widely to various contexts.
First, the vast majority of histone modifications occur either on the extended aminoterminal “tails” of histones, whereas a minority also occur on the histone octamer “core.”13 The core of the histone octamer wraps the DNA, whereas histone tails serve as platforms for the regulated binding of proteins, and covalent modifications can either enhance or deter binding of chromatin remodelers, chromatin modifiers, and transcription factors, and help to orchestrate protein associations during transcription (Fig. 12–2). For example, methylation on histone H3 (H3) H3K4me can deter interaction with DNMTs and therefore cause passive DNA demethylation; in contrast, H3K4me3 can facilitate interaction with RNA polymerase II (RNAP II) machinery.14 Second, histone modifiers are typically targeted by site-specific DNA binding proteins (see Fig. 12–2), which are themselves responsive to developmental and environmental/metabolic signaling. Third, some histone modifiers are targeted or regulated by other histone modifications, which underlies (in part) why certain sets of histone modifications are coincident in regions.13
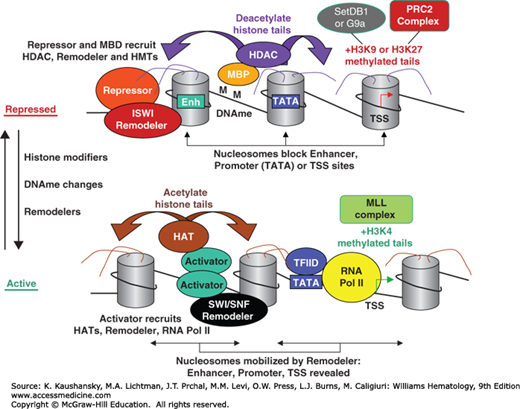
Figure 12–2.
Chromatin changes that accompany the transition from a repressed to an active state. Repression (top) is enforced by site-specific DNA-binding repressors which recruit factors such as histone deacetylates (HDACs), histone methyltransferases (HMTs; for H3K9me or H3K27me), and DNA methyltransferases (not shown), which are used to methylate the DNA (M). Methyl-binding domain proteins (MBDs) bind to DNA methylation (DNAme) and also recruit a similar set of chromatin modifiers. These repressors also recruit imitation SWI remodeler (ISWI)-family remodelers, which help position nucleosomes on important cis-controlling elements like enhancers (Enh), the TATA box, or the transcription start site (TSS). The transition from the repressed state to the active state involves the modification and repositioning of nucleosomes, as well as DNA demethylation by passive or active modes. Nucleosomes are modified by histone acetyltransferases (HATs), and activating HMTs (mixed-lineage leukemia [MLL] complex, specific for H3K4me). Such modifications are believed to be recognized by the bromodomains present on remodelers, which then mobilize modified nucleosomes, allowing the transcription machinery to bind. Components of the transcription machinery, such as TFIID, can also detect histone modifications.
A major concept in histone modification biology is dynamic reversibility, termed write, read, erase.1,15,16 “Writing” involves the enzymatic addition of a covalent modification to an amino acid, within a particular protein sequence context. “Reading” involves the ability of a second protein/domain to bind that modification, within a particular protein sequence context, defining the impact of the modification. “Erasing” involves the removal of the covalent modification, within a particular sequence context, regenerating the prior/initial state. These concepts are actually quite general, and can be applied widely in protein signal transduction biology, with this terminology simply having become popularized in the chromatin field. Nevertheless, these terms are quite useful for framing histone modification cycles that accompany transcription cycles. One illustrative example is the addition of histone acetylation by histone acetyltransferase (HAT) enzymes, the binding of acetylated histone tails by the bromodomain (present in SWI/SNF remodelers and certain chromatin modifiers),17,18 and the removal of acetylation by HDAC enzymes.19 Finally, although this chapter discusses histone modifications and modifiers, histone modifiers often also modify additional chromatin proteins, including proteins that contain “histone mimic” regions. Although beyond the scope of this chapter, those chromatin modifications often go through similar “write, read, erase” cycles to enable additional layers of protein recruitment and release within a chromatin process.









