Endocrine Tumors of the Pancreas
Maha T. Barakat
Houman Ashrafian
Stephen R. Bloom
Pancreatic endocrine tumors make up only 1% to 2% of all tumors of the pancreas but form an important group in that they tend to be more benign and treatable than the common exocrine tumors. Although the identification of the menin gene has provided some insights into the tumorigenesis of the rare familial form of pancreatic endocrine tumors, the heterogeneity of phenotypes and the absence of menin mutations in some cases remain unexplained. With the advent of technological advances facilitating the retrieval and analysis of large data sets, such as gene expression microarray profiling and multivariate analysis of complex traits (1), we are now poised to identify subsequent molecular events and use the information to prognosticate and risk stratify patients with the menin gene mutation. In addition, these techniques will permit comparison of the findings in patients with multiple endocrine neoplasia type 1 (MEN1) with those in patients with the more common sporadic form of disease in whom no menin mutation is apparent and to relate the pathogenesis of these potentially distinct but related tumor entities. To maximize the power of these basic science techniques, this analysis of the complex pathways in relation to tumor behavior will necessitate ever more rigorous clinical assessment of phenotype definitions. Indeed, gene-expression data have already been shown to provide invaluable prognostic information with regard to breast carcinoma (2,3), prostate carcinoma (4), and diffuse large B-cell lymphoma (5) and can predict imminent metastatic behavior in malignant melanoma (6).
Pancreatic endocrine tumors are derived from the diffuse neuroendocrine system of the gastrointestinal tract (7) and were formerly classified as apudomas (tumors of the a mine p recursor u ptake and d ecarboxylation system). These neuroendocrine cells can produce a myriad of hormones, many of which are also present in the brain. The hormones provide a complex network of communication, each having endocrine, paracrine, autocrine, or neurotransmitter functions crucial to the finely tuned functioning and regulation of the gastroenteropancreatic system.
INCIDENCE
Although the annual incidence of neuroendocrine pancreatic tumors is about 3.5 to 4 per million population, there is a vast discrepancy between the clinical incidence and that found post mortem, suggesting an abundance of asymptomatic disease. Pancreatic endocrine tumors have been detected in 0.3% to 1.6% of unselected autopsies in which only a few sections of the pancreas were examined, whereas they were found in up to 10% when the whole pancreas was examined systematically (8). Unlike neuroendocrine tumors of the gut, carcinoid tumors rarely occur in the pancreas and will not be covered in this chapter.
ETIOLOGY
The etiology of pancreatic endocrine tumors has many similarities to tumorigenesis in the better-studied colorectal carcinoma model. The similarities will be discussed in this section, and this
analogy will help in understanding the development of pancreatic endocrine tumors.
analogy will help in understanding the development of pancreatic endocrine tumors.
Genetic Model from Colorectal Tumorigenesis
It is becoming increasingly clear that tumorigenesis involves a series of multiple genetic alterations leading to activation of oncogenes and/or inactivation of tumor suppressor genes and ultimately to loss of apoptosis. The genetic basis of colorectal neoplasia has provided a salient model for this sequential process (9). The transition from healthy colonic epithelia through increasingly dysplastic adenoma to colorectal cancer involves the acquisition of mutations in specific genes, including APC, K-ras, DCC, and p53 [Fig. 70.1; data also from (10)]. Furthermore, although the total accumulation of genetic alterations is important, the exact sequence of events is critical for determining the phenotype, as illustrated by the early loss of Lkb1, rendering the cells resistant to oncogene-induced transformation, in Peutz-Jeghers syndrome (11).
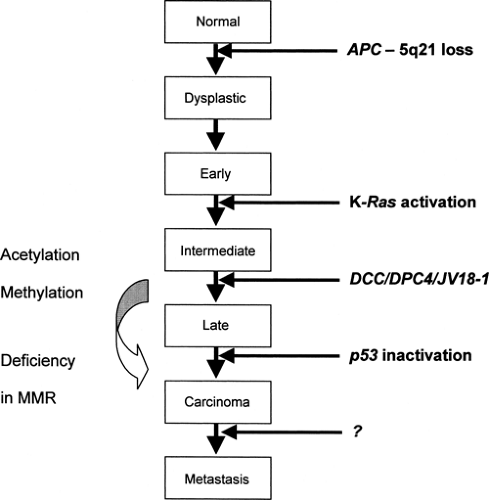 Figure 70.1. Genetic changes associated with colorectal tumorigenesis. Mutations in the adenomatous polyposis gene (APC) initiate tumorigenesis, and tumor progression results from mutations in the other genes indicated. MMR, mismatch pair. (Adapted from Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767; and Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;87:159–170; with additional data from Shannon BA, Iacopetta BJ. Methylation of the hMLH1, p16, and MDR1 genes in colorectal carcinoma: associations with clinicopathological features. Cancer Lett 2001;167:91–97.) |
Mutations in the adenomatous polyposis gene (APC) occur both in autosomal dominant familial adenomatous polyposis (FAP) and sporadically. The initiation of tumorigenesis requires a two-hit process, as outlined in Knudson’s model (12). In the case of germline mutations, the first hit to the APC gene is inherited with the germline, thus being identical in all cells. Alternatively, in somatic mutations the first hit can occur as a rare event in a particular somatic cell. A second hit in both germline and somatic cases inactivates the unaffected allele, resulting in loss of heterozygosity (LOH) and initiates tumorigenesis.
The APC gene, located on chromosome 5q21, encodes a 312-kDa tumor suppressor protein with many possible protein/ protein interactions, including binding sites for β-catenin, end-binding protein-1 (EB1), and axin (13). It is termed a “gatekeeper” gene, because in normal human colonic cells, the APC/axin protein complex facilitates the phosphorylation and subsequent degradation of β-catenin, thus preventing β-catenin-stimulated transcriptional activation and cell proliferation (Fig. 70.2) (14). It is therefore highly relevant that the majority of APC mutations in FAP result in the deletion of the carboxyterminal end, including the β-catenin and axin binding sites (15). This then initiates cell proliferation and tumorigenesis. Indeed, in addition to colorectal neoplasia, APC mutations have been associated with small-bowel tumors, desmoid tumors, and medulloblastomas.
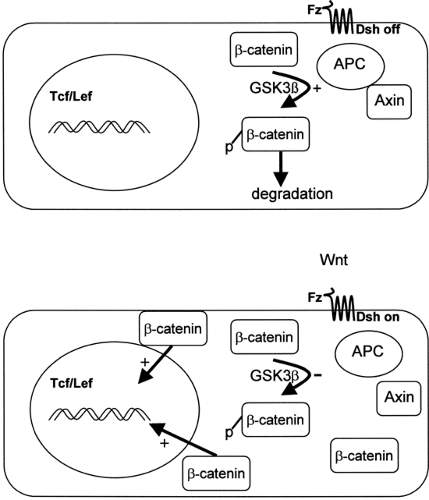 Figure 70.2. The APC/axin β-catenin “destruction pathway” and regulation of transcription. The APC/ axin complex modulates the activity of GSK3β (glycogen synthase kinase 3β) and facilitates the phosphorylation of β-catenin, leading to the degradation of β-catenin. When the cells are exposed to Wnt (wingless and integration activated gene product), which acts on cell surface receptors of the frizzled family (Fz), the APC/axin complex is inactivated by an unknown mechanism requiring disheveled protein (Dsh), and β-catenin accumulates in the cell. This enters the nucleus and interacts with Tcf/Lef (T-cell factor/lymphoid enhancer binding factor) DNA-binding protein to activate new gene expression. An analogous situation occurs with inactivating mutations in APC. APC, adenomatous polyposis. (Adapted from Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science 2000;287:1606–1609; and Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;87:159–170.) |
To a certain extent, there seems to be some correlation between genotype and phenotype in mutations affecting APC. Depending on the region of the gene affected, the resulting phenotype may be attenuated polyposis, classic polyposis, congenital hypertrophy of the retinal pigment epithelium (CHRPE), or Gardner syndrome (mandibular osteomas and desmoid tumors) (15). The resultant phenotype is determined by the functional consequences of the different mutations in APC and downstream modifying events.
Application of the Model to Pancreatic Endocrine Tumors
Although there is currently no direct evidence, it is likely that the process of tumorigenesis of pancreatic endocrine neoplasia follows the model of sequential accumulation of mutations, analogous to colonic neoplasia. In fact, there are many similarities between the two types of tumors. The familial form of pancreatic endocrine tumors is the autosomal dominant type MEN1, in which the other features include anterior pituitary tumors (27%); parathyroid hyperplasia (90%); and, more rarely, foregut carcinoid (14%), adrenocortical tumors (5%), and pheochromocytoma (<1%) (16). Nonendocrine tumors include angiofibroma (85%), collagenoma (70%), lipoma (30%), and
ependymoma (<1%). Pancreatic endocrine tumors can, however, also occur sporadically.
ependymoma (<1%). Pancreatic endocrine tumors can, however, also occur sporadically.
In 1988, Larsson and colleagues localized the MEN1 gene to chromosome 11q13 (17). Germline-inactivating mutations in this gene have been found in 95% of patients with familial MEN1, whereas in sporadic cases somatic mutations were found in a smaller proportion of patients: 21% of those with parathyroid adenoma, 33% with gastrinomas, 17% with insulinoma, 36% with bronchial carcinoids, and 50% to 70% with sporadic thymic and duodenal carcinoid (16,18,19,20,21). MEN1 is known to be a tumor suppressor, and it is likely that the transition from normal cells to neuroendocrine neoplasia in familial MEN1 requires a series of sequential genetic alterations starting with MEN1 [Fig. 70.3; data from (22,23,24,25,26)]. It is possible that, where there is no detectable MEN1 mutation (for instance, in the 5% of familial and in the majority of sporadic cases), the mutation cannot be detected given current techniques or that MEN1 is inactivated by mechanisms other than mutation, e.g., alterations in promoter methylation. It is interesting that the 5′-region of MEN1 contains a CpG-rich area, and hypermethylation of similar regions in the promoter of the VHL and the hMLH1 genes has been shown to be a frequent cause of somatic second hit in tumorigenesis in von Hippel-Lindau disease and colorectal carcinoma, respectively (27,28,29).
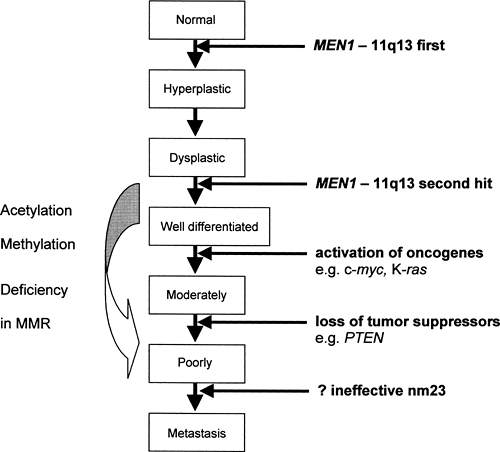 Figure 70.3. A model of genetic changes associated with pancreatic endocrine tumorigenesis. nm23 is a putative tumor metastasis suppressor. MEN1, multiple endocrine neoplasia type 1; MMR, mismatch pair; PTEN, phosphatase and tensin homologue. (Data from Crabtree JS, Scacheri PC, Ward JM, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Nath Acad Sci U S A 2001;98:1118–1123; Pannett AA, Thakker RV. Somatic mutations in MEN type 1 tumors, consistent with the Knudson “two-hit” hypothesis. J Clin Endocrinol Metab 2001;86:4371–4374; Pelengaris S, Khan M. Oncogenic co-operation in beta-cell tumorigenesis. Endocr Relat Cancer 2001;8:307–314; Wang L, Ignat A, Axiotis CA. Differential expression of the PTEN tumor suppressor protein in fetal and adult neuroendocrine tissues and tumors: progressive loss of PTEN expression in poorly differentiated neuroendocrine neoplasms. Appl Immunohistochem Mol Morphol 2002;10:139–146; and Yaguchi H, Ohkura N, Tsukada T, et al. Menin, the multiple endocrine neoplasia type 1 gene product, exhibits GTP-hydrolyzing activity in the presence of the tumor metastasis suppressor nm23. J Biol Chem 2002; 277:38197–38204.) |
Similar to colorectal neoplasia, the initiation of tumorigenesis from the MEN1 tumor suppressor gene requires a two-hit process [Knudson’s model (12,23)]. In germline mutations, the first hit to the MEN1 gene is inherited and only one more hit is required for loss of heterozygosity, whereas in somatic mutations of sporadic cases, two hits are required to result in tumorigenesis (17,30,31). In the mouse model with the homologue Men1 germline mutation, the remaining wild-type Men1 allele was retained until tumor formation, when somatic loss of this wild-type allele occurred in all tumors developed by the mice (22). The clinical features of familial MEN1 differ from somatic MEN1 mutations, in that age of onset of tumor expression is usually earlier (in keeping with the Knudson’s hypothesis) and that there is tumor multiplicity in terms of both multiple organs affected and also multiple tumors in one organ (16).
Menin, a 610-amino-acid protein, is the product of the MEN1 gene, and the native form has been shown to bind to and inhibit JunD, an activator protein-1 (AP-1) transcription factor (32). In addition, menin suppresses p65-mediated transcriptional activation on NF-κB sites (33), interacts with Smad3, facilitating transforming growth factor β (TGF-β)-mediated inhibition of cell growth (34), and may be important in the activity of a tumor metastasis suppressor nm23 (26), further suggestive of a tumor-suppressor role for menin (Fig. 70.4). Native menin has also been shown to inhibit insulin promoter activity and secretion and, when cotransfected into INS-1 insulinoma cells, results in inhibition of cell proliferation (35).
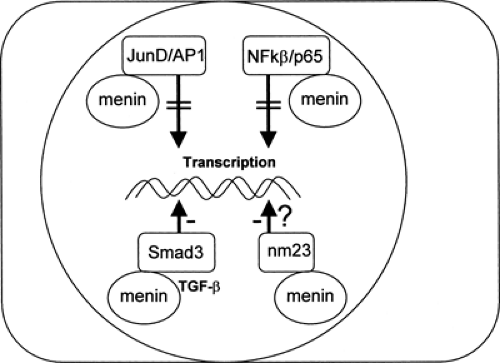 Figure 70.4. Interactions of native menin in its tumor-suppressor role. JunD is a member of the Jun family of proteins that dimerize with themselves or with Fos members to form the AP-1 (activator protein-1) transcriptional factor; menin binds JunD and inhibits JunD-mediated transcriptional activation. Menin also represses p65 (NF-κβ protein)-mediated transcriptional activation. Smad3, a downstream component of the transforming growth factor β (TGF-β) signaling pathway, interacts with menin to alter transcription, leading to inhibition of cell growth. Finally, in the presence of the putative tumor metastasis suppressor nm23, menin attains atypical GTPase activity, and this may affect transcription. (Data from Yaguchi H, Ohkura N, Tsukada T, et al. Menin, the multiple endocrine neoplasia type 1 gene product, exhibits GTP-hydrolyzing activity in the presence of the tumor metastasis suppressor nm23. J Biol Chem 2002;277: 38197–38204; Agarwal SK, Guru SC, Heppner C, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999;96:143–152; Heppner C, Bilimoria KY, Agarwal SK, et al. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene 2001;20: 4917–4925; and Kaji H, Canaff L, Lebrun JJ, et al. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc Natl Acad Sci U S A 2001;98:3837–3842.) |
Despite the similarities of the etiologies of pancreatic neuroendocrine tumors and the colorectal tumorigenesis model, several differences need to be highlighted. First, the slower rate
of growth of neuroendocrine tumors and in many cases a more benign progression exists, and this may be due intrinsically to more signaling blocks and to less exposure to carcinogens. Second, there seems to be very little in the way of genotype-phenotype correlation in MEN1 mutations (36). The development of, for instance, an insulinoma rather than a gastrinoma may be determined by local factors such as the number of pancreatic β-cells and their baseline proliferation rate. Most patients with familial MEN1 will develop nonfunctioning pancreatic tumors, whereas 40% will develop gastrinoma; 10%, insulinoma; and 2%, other functioning pancreatic tumors, such as glucagonoma, VIPoma (VIP, vasoactive intestinal polypeptide), and somatostatinoma (16).
of growth of neuroendocrine tumors and in many cases a more benign progression exists, and this may be due intrinsically to more signaling blocks and to less exposure to carcinogens. Second, there seems to be very little in the way of genotype-phenotype correlation in MEN1 mutations (36). The development of, for instance, an insulinoma rather than a gastrinoma may be determined by local factors such as the number of pancreatic β-cells and their baseline proliferation rate. Most patients with familial MEN1 will develop nonfunctioning pancreatic tumors, whereas 40% will develop gastrinoma; 10%, insulinoma; and 2%, other functioning pancreatic tumors, such as glucagonoma, VIPoma (VIP, vasoactive intestinal polypeptide), and somatostatinoma (16).
Similarly, there is a wide spectrum in the aggressiveness of different pancreatic neuroendocrine tumors. The tumor phenotype will be determined by many factors (the particular MEN1 mutation itself and many other downstream effects). As previously mentioned, familial MEN1 tends to result in a more aggressive tumor phenotype (earlier onset and multiplicity) compared with somatic mutations. The loss of expression of the phosphatidylinositol 3′-phosphatase and tumor suppressor gene PTEN (phosphatase and tensin homologue) on chromosome 10 has been shown in 54% (7 of 13) of poorly differentiated neuroendocrine tumors, whereas the two moderately differentiated neuroendocrine tumors in the series retained PTEN immunostaining (25). PTEN may be only one of many downstream factors that determine phenotype.
PHYSIOLOGY
There is a complex relationship between cells and enteric neurons in the gastroenteropancreatic system. This is orchestrated by a vast number of regulatory peptides secreted from neuroendocrine cells, which act in an autocrine, paracrine, and endocrine manner as well as an neurocrine manner, in which the peptides behave as neurotransmitters and neuromodulators (7,37,38,39,40).
CLINICAL FEATURES
Despite the plethora of known gut hormones, it is possible that some still cannot be detected by current techniques. When a neuroendocrine tumor is diagnosed histologically, if no circulating gut hormone is found, it is termed “nonfunctioning.” However, it may just be that the secreted hormone is as yet undetectable. The patients tend to be asymptomatic initially and to develop symptoms from tumor bulk at later stages of disease progression (41). On the other hand, for those “functioning” tumors that secrete detectable hormones (e.g., insulin or gastrin), the clinical picture is very much dependent on the nature of the secreted hormone, each giving a defined syndrome [Table 70.1; data from (40,42,43,44,45)], rather than on tumor bulk. Indeed, it is common for more than one hormone to be co-secreted at presentation or even for a tumor to dedifferentiate and start producing multiple hormones. This is particularly the case with MEN1. Genetic testing for MEN1 is now readily available, and screening of family members is recommended once a germline menin mutation is found.
TABLE 70.1. Tumor Syndromes of Pancreatic Endocrine Tumors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
IMMUNOCYTOCHEMICAL AND BASELINE BIOCHEMICAL MARKERS
The neuroendocrine tumors share certain general immunocytochemical markers, including the secretory granules chromogranins, neuron-specific enolase (NSE), pancreatic polypeptide, human chorionic gonadotropin (hCG) subunits, and peptide histidine-methionine (46,47,48). Chromogranins A, B, and C (CgA, CgB, CgC) are located alongside specific hormones in large
dense-core vesicles of neuronal and neuroendocrine cells, with CgA being the most widely distributed (49). With the exceptions of the lactotrophs of the pituitary and some pancreatic β-cell tumors, which show positive staining for CgB, CgA-positive tumors include anterior pituitary, parathyroid, medullary thyroid, gastroenteropancreatic, ectopic ACTH (adrenocorticotropic hormone), ganglioneuromas, neuroblastomas, pheochromocytoma, and small-cell lung and prostate tumors.
dense-core vesicles of neuronal and neuroendocrine cells, with CgA being the most widely distributed (49). With the exceptions of the lactotrophs of the pituitary and some pancreatic β-cell tumors, which show positive staining for CgB, CgA-positive tumors include anterior pituitary, parathyroid, medullary thyroid, gastroenteropancreatic, ectopic ACTH (adrenocorticotropic hormone), ganglioneuromas, neuroblastomas, pheochromocytoma, and small-cell lung and prostate tumors.
Some of the above-mentioned tissue markers can be detectable in plasma. Plasma CgA is elevated in 94% of pancreatic endocrine tumors, whereas pancreatic polypeptide is elevated in 74% (50). CgA, however, seems to be the best marker for metastatic carcinoid, whereas plasma CgB appears better for detecting benign insulinomas. A fragment of CgB termed GAWK (CgB-420-493) is a 74-amino-acid peptide originally isolated from human pituitaries, and high plasma concentrations are found in various neuroendocrine tumors, including pancreatic, pheochromocytoma, medullary carcinoma of the thyroid, and ACTH-producing lung tumors. GAWK is particularly elevated in pancreatic endocrine tumors, making it a useful marker (51). In another study of pancreatic endocrine tumors, pancreastatin was elevated in 33% of patients, whereas GAWK was elevated in 50% (52).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


