- Endocrine causes of diabetes are mainly a result of an excess of hormones that are counter-regulatory to insulin, and act by inhibiting insulin secretion and/or action.
- Acromegaly is almost always secondary to growth hormone-secreting adenomas of the anterior pituitary somatotrophs and disturbs glucose homeostasis in up to approximately 50% of patients.
- Cushing syndrome is caused by excessive levels of glucocorticoids and disturbs glucose homeostasis to some degree in over 50% of cases.
- Pheochromocytoma is a tumor of the chromaffin cells, which in 90% of cases is located in the adrenal medulla and causes hyperglycemia in approximately 50% of cases.
- Glucagonoma and somatostatinoma are rare islet cell tumors that produce hormones that inhibit the secretion and action of insulin.
- Thyrotoxicosis commonly causes mild glucose intolerance, but overt diabetes only occurs in a tiny minority.
- Other endocrinopathies such as primary aldosteronism and primary hyperparathyroidism can disturb glucose homeostasis.
- Polycystic ovarian syndrome occurs in 5–10% of women of reproductive age and associates with some degree of glucose intolerance or diabetes resulting from insulin resistance in approximately 50% of cases.
Introduction
The primary focus of this chapter is on those endocrine disorders that cause hyperglycemia and where effective treatment of the endocrinopathy can be expected to normalize the blood glucose concentration. These conditions mostly reflect excessive secretion of “counter-regulatory” hormones, the metabolic actions of which oppose those of insulin by inhibiting its secretion, action, or both.
Acromegaly
Etiology, incidence and clinical features of acromegaly
Acromegaly comprises a constellation of symptoms and signs caused by excessive growth hormone (GH) secretion that leads to bony and soft tissue overgrowth accompanied by cardiovascular and metabolic pathology (Figure 17.1a; Table 17.1) [1]. It affects approximately 60 people per million [2] and, in 99% of cases, is caused by a pituitary adenoma, most commonly larger than 1 cm in diameter (a “macroadenoma”; Figure 17.1b). A tiny minority of cases are caused by excessive secretion of GH-releasing hormone (GHRH) from a hypothalamic gangliocytoma or a carcinoid tumor of the lung or pancreas [1]. A small percentage of acromegaly occurs within the wider endocrine syndrome of multiple endocrine neoplasia type 1 (MEN1) caused by mutations in the tumor suppressor gene, MENIN [3]. MEN1 can also include glucagonomas and somatostatinomas, both of which are separately capable of causing secondary diabetes. Commonly, acromegaly has been present for a decade prior to diagnosis [4]. This long-standing hypersecretion of GH provides the time necessary for the characteristic external features of the disorder to develop (Figure 17.1a; Table 17.1).
Figure 17.1 Acromegaly in a patient found to have a random blood glucose level of 13 mmol/L during preparation for sinus surgery. Features included: (a) the characteristic facial appearance; (b) a large adenoma (arrow) extending up to but not in contact with the optic chiasm demonstrated by magnetic resonance imaging (R, right; L, left). Following successful trans-sphenoidal removal of the tumor, glucose tolerance returned to normal.
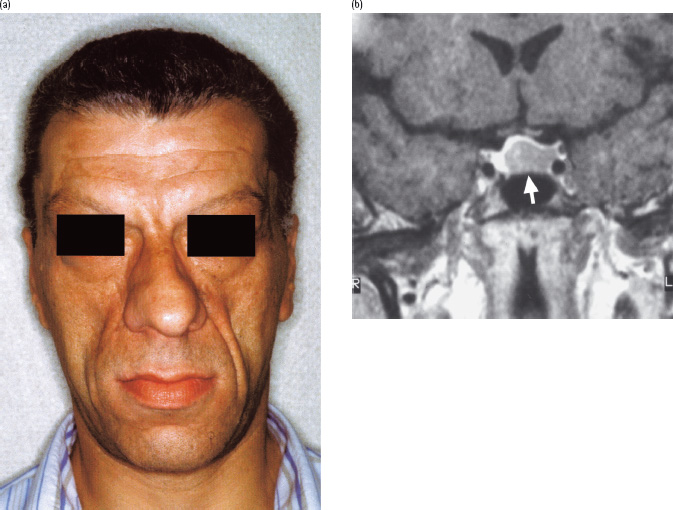
Table 17.1 Clinical features of acromegaly.
| Musculoskeletal |
| Protruding mandible (prognathia) with lower teeth separation |
| Big tongue (macroglossia) |
| Enlarged forehead (frontal bossing) |
| Large hands and feet (carpal tunnel syndrome, tight rings, increasing shoe size) |
| Osteoarthritis from abnormal joint loading |
| Increased stature (gigantism; if GH excess occurs prior to epiphyseal closure) |
| Skin |
| Irritating, thickened, greasy (increased sebum production) |
| Excessive sweating |
| Cardiovascular |
| Dilated cardiomyopathy causing cardiomegaly and cardiac failure |
| Hypertension |
| Metabolic |
| Impaired glucose tolerance or potentially diabetes |
| General |
| Headaches |
| Tiredness, often very disabling and lowers quality of life/ability to work |
| Local tumor effects |
| Compression of the optic chiasm (superior tumor growth) or cranial nerves III, IV and/ or VI (lateral tumor growth into cavernous sinus) |
GH, growth hormone.
Features of disturbance to glucose tolerance in acromegaly
Glucose intolerance or overt diabetes is common in acromegaly because of the direct hyperglycemic effects of GH excess (Figure 17.2). Overt diabetes has been reported in a range of 19–56% of patients with acromegaly, while impaired glucose intolerance (IGT) affects 16–46% [5–8]. Diabetes is most frequent in patients with higher GH levels [9,10]. There is also a correlation between serum insulin-like growth factor I (IGF-I), and both fasting and post-prandial glucose. In fact, placing age-adjusted IGF-I values into rising quartiles correlated very accurately to decreasing insulin sensitivity such that serum IGF-I levels could predict insulin sensitivity more accurately than either random GH levels or the nadir value following glucose tolerance testing [11].
Figure 17.2 Mechanisms of hyperglycemia and diabetes in acromegaly. Diabetes develops if β-cells fail to compensate for the increased demand for insulin. IGT, impaired glucose tolerance; NEFA, non-esterified fatty acid.
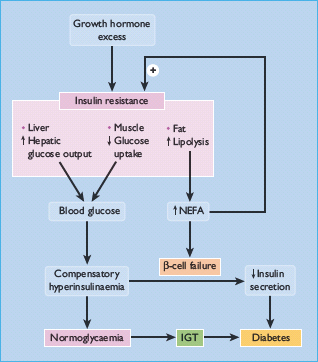
Clinically, diabetes in acromegaly usually resembles the type 2 form (T2DM), with most patients not needing insulin therapy but being maintained with oral hypoglycemic agents or diet alone [9]. GH can induce insulin resistance when infused into normal subjects [12,13]. and insulin resistance is a consistent feature of people with acromegaly [14,15]. Insulin action is impaired in both the liver and extrahepatic tissues, with decreases in both the suppression of hepatic glucose production and in insulin-dependent glucose disposal [13,15]. For instance, impairment of insulin-mediated activation of glycogen synthase has been demonstrated in skeletal muscle [16–18]. Insulin resistance may also be exacerbated by the lipolytic action of GH, generating non-esterified fatty acids (NEFAs) which act on the liver to increase glucose production and in muscle to inhibit glucose utilization (via the “glucose-fatty acid” cycle) (Figure 17.2). Where pancreatic compensation is adequate, an exaggerated insulin secretory response creating hyperinsulinemia can counterbalance the insulin resistance and maintain euglycemia. Similar to the natural history of deteriorating blood glucose control in T2DM, eventually β-cell compensation fails, the insulin response is impaired and hypergylcemia ensues [14]. This has been termed the Starling curve of the pancreas (see Figure 11.1).
Diagnosis and treatment of acromegaly
GH release is normally pulsatile with intervening periods of low level or undetectable hormone, whereas secretion from pituitary adenomas is autonomous. Therefore, the diagnosis can be deduced from a series of random serum measurements where GH is consistently detected. An alternative, better diagnostic test takes advantage of the negative feedback on GH secretion by glucose. A failure of serum GH to suppress to below 2 mU/L (approximately 1 μg/L) within 3 hours of 75 g oral glucose is diagnostic of acromegaly. A further option, particularly suited as the initial screening test of outpatients, is measurement of serum IGF-I, which if raised above sex- and age-matched controls is diagnostic of acromegaly, unless there is a possibility of GH abuse [1]. It is noteworthy that the diagnosis of acromegaly can be difficult in patients with type 1 diabetes mellitus (T1DM) where GH hyper-secretion is observed. Whereas this might compromise the use of serum GH as a diagnostic biomarker of a somatotroph adenoma, IGF-I values tend to be low in poorly controlled T1DM indicative of the state of GH resistance. Thus, raised serum IGF-I values remain a confident predictor for a diagnosis of acromegaly in people with T1DM. Having diagnosed acromegaly, magnetic resonance imaging (MRI) of the anterior pituitary defines the extent of the adenoma (Figure 17.1b).
Based on this investigation, symptoms, patient wishes and co-morbidity, and GH levels, treatment can be either surgical, medical or radiotherapy (Table 17.2) [1,19]. When the tumor is a macroadenoma, especially if it extends beyond the pituitary fossa, curative surgery becomes unlikely or impossible; however, surgery can be useful for “debulking” and rapid lowering of serum GH levels in patients who are highly symptomatic, or where the tumor has encroached upon the optic chiasm and affected visual fields. The standard approach is trans-sphenoidal, either via the nostril or from behind the upper lip. Once the sphenoid sinus has been traversed and midline access to the sella turcica gained, tumor is removed from the anteroinferior aspect causing the residual tissue to drop back down into or towards the pituitary fossa. Tumor beyond the fossa, in locations such as the cavernous sinus, cannot be approached directly, hence the reason why surgery for large tumors is not anticipated to be curative [19]. Conversely, cure can be commonly achieved for over 50% of microadenomas (<1 cm diameter). The reported ranges vary widely according to expertise.
Table 17.2 Treatment of acromegaly (Adapted from Holt and Hanley [130].)
| Advantages | Disadvantages |
| Trans-sphenoidal surgery | |
| Rapid effect | Invasive and requires general anesthetic |
| Can restore vision in optic nerve compression | Non-curative for large extrasellar tumors |
| Might be curative if complete resection | |
| Somatostatin analog drugs | |
| Non-invasive | Monthly intramuscular injection |
| May shrink tumor | Expensive |
| Decreases GH in ∼60% of patients | Gastrointestinal side effects (commonly diarrhea) |
| Unlikely to be curative (i.e. continuous therapy needed) | |
| Pegvisomant | |
| Non-invasive | Expensive |
| Blocks GH action | GH concentrations remain elevated |
| Radiotherapy | |
| Non-invasive | Slow to act – may take up to 10 years |
| Likely to shrink tumor | Standard external three-beam |
| Likely to reduce GH levels | radiotherapy likely to cause |
| Might be curative | hypopituitarism by destroying other pituitary cell types |
GH, growth hormone.
Medical therapy, most commonly using somatostatin analogs, is effective at both lowering GH levels and shrinking the tumor volume [1,20,21]. Approximately 60% of patients respond to somatostatin analogs because of the presence of predominantly type 2 and type 5 somatostatin receptors on the tumor cell surface [1,21]. The analogs can be administered subcutaneously; however, once it is clear that they are tolerated, the most common formulation is month-long intramuscular depot preparations. They can be used either prior to surgery, with the goal of operating on a shrunken tumor, on a long-term basis in place of surgery or post-surgery where GH levels have not been normalized.
Some GH-secreting adenomas co-express dopamine receptors more characteristic of prolactinomas [21]. Indeed, 25% of cases of acromegaly show raised levels of both prolactin and GH possibly indicating a tumor cell phenotype more consistent with the somatomammotroph from which it is thought that somatotrophs (GH secretion) and lactotrophs (prolactin secretion) terminally differentiate. In these instances, dopamine agonists, as used in hyperprolactinemia, can be useful, especially as they can be administered orally, and allow reduction in dosage of the more expensive intramuscular depot somatostatin analogs. This opportunity to use lower doses of somatostatin analogs may also lessen their side-effects, such as gastrointestinal disturbance (most commonly diarrhea) and gallstones. It has recently been questioned, however, whether commonly used ergot alkaloid derived dopamine agonists, such as cabergoline, cause fibrotic side-effects, especially involving heart valves [22,23]. Despite concerns from regulatory agencies, the prevailing view from endocrinologists is that the doses of these agents used to treat endocrine disorders (compared with the therapeutic regimens in Parkinson disease) are not problematic. In any case, alternative non-ergot derived agents, such as quinagolide, are available. Bromocriptine is less commonly used because of the almost inevitable side-effects of nausea. Responsiveness to both dopamine and somato-statin analog therapy can be easily assessed at the start of treatment by hourly measurement of serum GH over 8 hours following the administration of a test dose of each agent given sequentially on two consecutive days.
The last decade has seen the appearance of a new clinical agent that blocks GH action. GH induces signal transduction via binding to its receptor as a dimer. Pegvisomant has been developed as a GH antagonist by preventing this dimerization and thus inhibiting GH action. This leaves elevated GH levels from the somatotroph adenoma but, nevertheless, pegvisomant is effective at reversing the clinical features of acromegaly [1,24]. The major problem with its use in many countries has been its prohibitive cost. Concern over tumor growth because of loss of negative feedback (a scenario akin to Nelson syndrome following bilateral adrenalectomy in Cushing disease) seems unfounded [1,24].
Radiation therapy is most commonly administered as conventional three-field external beam radiotherapy [1]. It is effective at lowering GH and IGF-I levels [25]. This approach delivers approximately 4500 Gy to the pituitary region with the total dose calculated such that the optic chiasm receives less than 8 Gy. An alternative is stereotactic radiotherapy (also known as γ-knife therapy or radiosurgery), which by using more sources can focus a higher concentration of radiation to a defined area of tumor. Whereas the latter modality allows greater preservation of adjacent normal pituitary tissue, the former approach is a more all-encompassing strategy to ensure tumor destruction, albeit with a higher post-therapy incidence of hypopituitarism. The choice is important as there is evidence that pituitary radiotherapy is associated with increased morbidity and mortality from subsequent cerebrovascular disease meaning that repeat therapy is not undertaken lightly [26].
Outcome of acromegaly and disturbance to glucose tolerance
Unless there are cogent reasons against, attempts should be made to normalize GH excess in acromegaly to decrease the morbidity and mortality associated with the disorder [1,26]. Curative treatment is best defined by a nadir serum GH of less than 2 mU/L upon 75 g oral glucose challenge. Glucose tolerance improves and insulin levels decrease after successful treatment by pituitary surgery and irradiation [27,28]. Hyperglycemia may worsen in a few patients, presumably those with a stronger underlying tendency to T2DM, when treated with somatostatin analogs because these drugs also suppress insulin secretion [29,30]. In the longer term, somatostatin analogs tend to improve glucose tolerance [31,32]. Insulin sensitivity is also improved with pegvisomant therapy in patients with both glucose intolerance and diabetes [33–36]. If hyperglycemia persists after serum GH levels have been normalized then the patient should be considered as having T2DM. One caveat to this is the possibility of GH deficiency as part of hypopituitarism post-surgery or radiotherapy [14]. GH deficiency causes centripetal fat deposition, which would itself cause or accentuate insulin resistance and potential loss of euglycemia.
Generally, diabetic complications are considered rare in acromegaly. Nevertheless, it is worth considering GH action in the context of both macrovascular and microvascular diabetic complications. Untreated GH excess increases cardiovascular morbidity and mortality and, potentially, abnormalities of glucose metabolism occurring in acromegaly could contribute to hypertension which affects over 50% of patients with acromegaly [5]. GH has been linked to proliferative diabetic retinopathy ever since resolution of diabetic retinopathy was noted in a woman who developed rapid-onset pan-hypopituitarism [37,38], so much so that hypophysectomy was used as treatment prior to the advent of laser photocoagulation [38]. This view is supported by the observations that patients with diabetes who are GH-deficient rarely develop retinopathy [39]. and an experimental study in mice demonstrated that inhibition of GH secretion ameliorated diabetic retinopathy [40]. Patients with diabetes who are acromegalic, however, do not show an increased incidence of diabetic retinopathy [41,42]. Data on the progression of diabetic retinopathy treated with either pegvisomant or somatostatin analog are contradictory, and the outcome of larger trials are awaited [38].
Effects of diabetes on GH–IGF-I axis
Dysregulation of the GH–IGF-I axis has been well documented in T1DM. The main disturbances include increased GH secretion (Figure 17.3), paradoxically associated with decreased serum IGF-I levels [43]. GH secretory pulses are larger and more frequent and total 24 hour serum and urinary GH levels are elevated. High circulating GH levels are most obvious during periods of poor diabetic control, and return towards normal with improved control. Pulses of GH secretion during sleep in the early hours of the morning lead to insulin resistance, which manifests itself before breakfast and is largely responsible for the “dawn phenomenon” of fasting hyperglycemia [44]. This physiologic effect, along with that caused by exercise and GHRH, is exaggerated in patients with T1DM, especially those who are poorly controlled. GH secretion accounts for much of the decreased insulin sensitivity observed during normal puberty, as well as the deterioration in glycemic control at this time in adolescents with T1DM. Despite GH hypersecretion, levels of IGF-I – the principal mediator of GH activity – are inappropriately low, indicating a state of GH resistance (Figure 17.4). Hepatic resistance to GH has been attributed both to decreased number of GH receptors and to post-receptor defects [43]. Circulating levels of GH-binding protein (GHBP) are also decreased in T1DM. Administration of recombinant human IGF-I (rhIGF-I) as an adjunct to insulin has been demonstrated to reverse GH hypersecretion and improve glycemic control, while reducing insulin requirements. Co-administration of IGF-binding protein 3 (IGFBP-3) with IGF-I has similar beneficial effects on carbohydrate metabolism, and also avoids side effects of IGF-I, notably edema, headache, retinal edema and jaw pain [45]. Recombinant IGF-I has also been used to treat states of severe insulin resistance in which the insulin receptor is functionally impaired, such as the spectrum from Donohue to Rabson-Mendenhall syndrome (see Chapter 15) [46]. In such patients, rhIGF-I can be used to treat ketoacidosis, which is the major cause of death and may fail to respond even to massive doses of insulin.
Figure 17.3 Increased growth hormone (GH) secretion in type 1 diabetes. Note the marked hypersecretion during sleep in the early hours of the morning. Reproduced from Hansen et al. [127], with permission from Wiley-Blackwell.
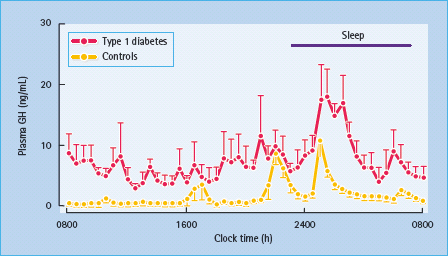
Figure 17.4 Mechanisms of growth hormone (GH) hypersecretion in diabetes. Insulin – like growth factor 1 binding protein 1 (IGFBP-1) binds and reduces the bioavailability of insulin – like growth factor I (IGF-I), which normally decreases GH secretion by negative feedback inhibition on the hypothalamus and pituitary; IGFBP-1 expression is inhibited by insulin.
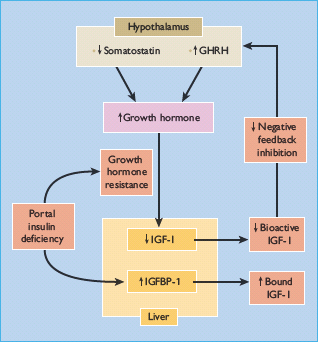
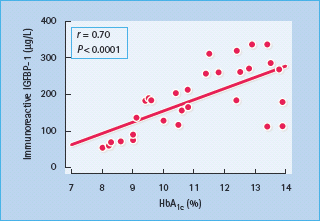
Abnormalities of IGFBP may also alter the regulation of the GH–IGF-I axis in diabetes (Figure 17.4). Six IGFBPs have been identified in humans, the two most relevant being IGFBP-1 (molecular weight 25 kDa), whose synthesis by hepatocytes is inhibited by insulin but independently of GH, and IGFBP-3 (molecular weight 44 kDa), which is GH-dependent [47]. IGFBP-1 levels are increased in T1DM as a result of portal and hepatic insulin deficiency (Figure 17.4) [43], and increase with worsening insulin deficiency and rising HbA1c concentrations (Figure 17.5). Increased IGFBP-1 levels have been shown to inhibit IGF-I bio-activity. Portal insulin deficiency in T1DM can thus account for GH resistance, through downregulation of hepatic GH receptors and increased IGFBP-1 levels, both reducing IGF-I production and bioactivity [48]. Decreased IGF-I levels in turn cause GH hypersecretion via reduced negative feedback at the hypothalamus and pituitary, and this exacerbates insulin resistance, thus establishing a vicious circle of raised GH and poor glycemic control (Figures 17.2 and 17.3).
Figure 17.5 Correlation between serum immunoreactive insulin-like growth factor 1 binding protein 1 (IGFBP-1) and glycated hemoglobin (HbA1c) concentrations in 48 patients with type 1 diabetes mellitus. Reproduced from Langford and Miell [128], with permission from the European Journal of Clinical Investigation
Another consequence of IGF-I deficiency is impaired growth at puberty [48]. Paradoxically, children with new-onset diabetes tend to be taller, especially if the disease develops several years before puberty; increased GH and insulin levels during the pre-clinical evolution of the disease are a possible explanation. Once diabetes is established, growth may slow, particularly before the age of 10 years and if glycemia is poorly controlled (Figure 17.6). The pubertal growth spurt may be blunted and/or delayed, especially in girls, and this may lead to a reduction in final height [48]. Growth failure in adolescents with T1DM is rare nowadays, possibly because of improved management and monitoring of diabetes. Indeed, intensified insulin treatment has been shown to raise IGF-I levels, in parallel with an increase in growth velocity [48]. Finally, growth failure may be associated with truncal obesity, hepatomegaly (secondary to glycogen and/or triglyceride deposition) and sexual infantilism in the Mauriac syndrome [49]. This condition was reported in children with poor glycemic control and excessively high insulin dosages, but is now rare.
Figure 17.6 Growth failure in a boy with diabetes. Height fell progressively below the centiles (97th centile at diagnosis) and the pubertal growth spurt was delayed, resulting in a lower fi nal height than predicted.
Cushing syndrome
Etiology, incidence and clinical features of Cushing syndrome
Cushing syndrome comprises a constellation of symptoms and signs caused by excessive levels of glucocorticoid that leads to a characteristic appearance accompanied by metabolic and cardiovascular pathology (Figure 17.7; Table 17.3) [50–52]. It occurs most commonly as a side effect of synthetic glucocorticoids administered exogenously for conditions such as rheumatoid arthritis or reversible airways disease. Endogenous Cushing syndrome arises in approximately two-thirds of cases from adrenocorticotropin (ACTH) secreting corticotroph adenomas of the anterior pituitary which affect 5–10 individuals per million. In one-fifth of cases, the cause is a glucocorticoid-secreting tumor of the adrenal cortex and in one-tenth of cases is secondary to syndromes of ectopic ACTH secretion, most commonly from small cell carcinoma of the lung or, more rarely, carcinoid tumors [50–52]. Cushing syndrome is more common in women than men, with a greater predilection for corticotroph adenomas as the underlying pathology. Prolonged excessive levels of cortisol, the major glucocorticoid in humans, either directly from an adreno-cortical tumor or from excessive ACTH causing bilateral hyper-functional adrenal cortices, causes the characteristic external features of the disorder to develop and results in excess cardiovascular morbidity and mortality (Fig. 17.7a; Table 17.3) [50–52].
Figure 17.7 Cushing syndrome in a 53-year-old hypertensive man presenting with diabetes that required insulin to control hyperglycemia. Features included back pain from osteoporotic vertebral collapse, proximal myopathy that prevented him from climbing stairs, characteristic rounded facies (a) and truncal obesity with violaceous striae (c). After removal of the 10-mm corticotroph pituitary adenoma, his facial appearance returned to normal (b), as did blood pressure and blood glucose.
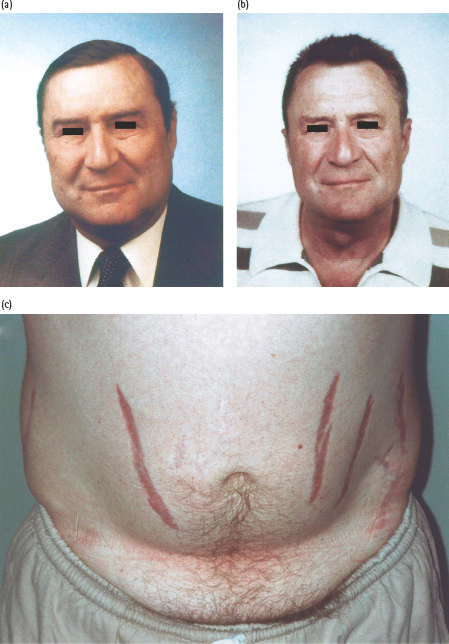
Table 17.3 Clinical features of Cushing syndrome.
| Easily bruised, thin skin; poor wound healing |
| Striae (purple or “violaceous” rather than white) |
| Thin (osteoporotic) bones that easily fracture |
| Glucose intolerance/diabetes mellitus |
| Central obesity, characteristic rounded facies, “buffalo” hump |
| Susceptibility to infection |
| Predisposition to gastric ulcer |
| Hypertension |
| Disturbance of menstrual cycle; symptoms overlap with PCOS |
| Mood disturbance (depression, psychosis) |
PCOS, polycystic ovarian syndrome.
Features of disturbance to glucose tolerance in Cushing syndrome
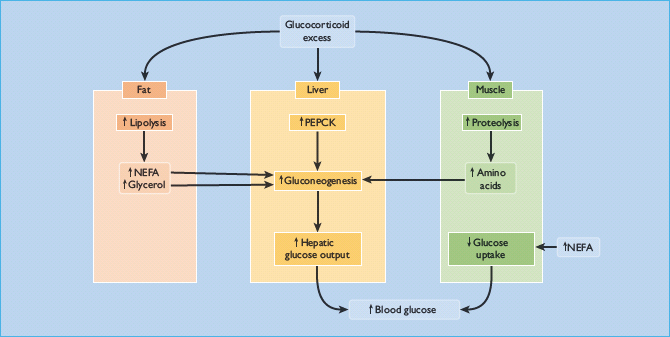
Impairment of glucose tolerance is observed in 30–60% of cases [7,50,53]. Overt diabetes occurs in 20–50%, arguably in patients predisposed to diabetes (e.g. those with a family history of the disorder) [7,50,53]. It resembles T2DM because glucocorticoid excess causes hyperglycemia primarily by inducing insulin resistance, reflected by hyperinsulinemia [53]. Insulin-stimulated glucose uptake and utilization by peripheral tissues are both reduced, while hepatic glucose production is greatly increased through stimulation of gluconeogenesis [50,54]. This results from the direct activation of hepatic gluconeogenic enzymes such as phosphoenolpyruvate carboxykinase (PEPCK), and an increasing supply of glucogenic substrates (amino acids and glycerol) generated by muscle proteolysis and peripheral adipose tissue lipolysis (Figure 17.8) [55]. Glucocorticoids also have permissive effects on the gluconeogenesis induced by epinephrine (adrenaline) and glucagon. Besides enhancing hepatic gluconeogenesis, glucocorticoids also increase hepatic glycogen storage [55].
Figure 17.8 Mechanisms of hyperglycemia and diabetes in Cushing syndrome. NEFA, non-esterified fatty acids; PEPCK, phosphoenolpyruvate carboxykinase.
With endogenous causes of Cushing syndrome, the hyperglycemia can sometimes be effectively treated with sulfonylureas, but many cases require insulin therapy [56]. For Cushing syndrome caused by exogeneous drugs, treatment can be tailored from knowledge of the timing and half-life of the glucocorticoid drug as a predictable period of hyperglycemia follows. For instance, prednisolone administered for reversible airways disease at breakfast will generate elevated glucocorticoid levels throughout the remainder of the day with levels falling in the evening. This profile makes the choice of an intermediate acting insulin administered at breakfast appropriate (see Chapter 27).
Diagnosis and treatment of Cushing syndrome
For exogenously administered glucocorticoids, the diagnosis and treatment of Cushing syndrome are straightforward: wherever possible remove or reduce the offending medication. The major challenge in diagnosing Cushing syndrome resulting from endogenous causes is frequently one of simply considering the condition as the underlying cause of otherwise very common symptoms such as tiredness and weight gain. Avoiding this pitfall is aided by thorough clinical examination when more specific signs may be detected, for instance violaceous stretch marks (called “striae”; Figure 17.7c) or proximal myopathy [50–52]. This issue may be particularly pertinent to the T2DM clinic, with some evidence that occult Cushing syndrome may affect 2% of patients [57].
The first goal is to establish the presence of either autonomous secretion or excessive levels of cortisol [50–52]. Several screening tests with high sensitivity have been designed (Table 17.4). Circulating cortisol is relatively high during the day and low at bedtime. Maintained daytime levels at night indicate excess. This can be tested by measuring midnight serum cortisol, although this requires prior acclimatization of inpatients for at least 24 hours and quiet surroundings as cortisol levels rise with minimal stress. Where assays have been validated, bedtime salivary cortisol measurement can be very useful as patients can post samples to the laboratory from home. Levels are approximately 10% of those in serum so care must be taken to avoid contamination with blood (e.g. from brushing teeth). The “low dose” dexamethasone suppression tests interrogate the potential loss of physiologic negative feedback at the corticotroph leading to autonomous ACTH secretion. The formal 48-hour test is marginally more specific [51]; however, it is also more inconvenient to perform in the outpatient setting compared with the overnight 1 mg suppression test. Caution needs to be exercised in patients where dexamethasone metabolism is enhanced (e.g. by antiepileptic medication) as this can lead to false-positive results. An alternative screening test examines total excretion of cortisol in a 24-hour urine collection, commonly on several occasions, as an integrated assessment of one day’s adrenocortical function. It is usual for endocrinologists to apply more than one of these various screening tests, which, if failed, provides proof of excess glucocorticoids and a diagnosis of Cushing syndrome.
Table 17.4 Diagnosis of Cushing syndrome from endogenous causes.
| Test | Interpretation |
| Screening tests to diagnose Cushing syndrome | |
| Midnight serum cortisol measurement or bedtime salivary cortisol assay | Maintained daytime levels at night-time indicates autonomous cortisol production of Cushing syndrome |
| 1 mg overnight DST. Oral dexamethasone taken at midnight | Serum cortisol > 50 nmol/L at following 9 AM consistent with Cushing syndrome |
| Formal low-dose DST (0.5 mg × 8 doses 6-hourly ending at 3 AM) | Serum cortisol > 50 nmol/L at following 9 AM consistent with Cushing syndrome |
| 24-hour urinary free cortisol measurement | Elevated values support diagnosis of Cushing syndrome |
| Tests to localize cause of cortisol excess | |
| Serum ACTH measurement | If suppressed, indicates autonomous adrenocortical overproduction of cortisol (e.g. an adrenocortical adenoma) |
| If serum ACTH detectable: | |
| High dose dexamethasone suppression test (2 mg × 8 doses 6-hourly ending at 3 AM) | > 50% suppression of 9 AM serum cortisol from pre-to post-test indicates anterior pituitary source; < 50% suppression indicates extrapituitary “ectopic” source of ACTH |
| Bilateral IPS sampling | Gradient from IPS to periphery of > 2:1 supports anterior pituitary source (test can also incorporate CRH administration; see text) |
| MRI (can include gadolinium enhancement) | Should only be considered once biochemical evidence of pituitary source obtained. Helpful for surgeon in planning trans-sphenoidal surgery |
ACTH, adenocorticotropic hormone; CRH, corticotropin releasing hormone; DST, dexamethasone suppression test; IPS, inferior petrosal sinus; MRI, magnetic resonance imaging.
Several endocrine tests have been developed to differentiate the various etiologies of endogenous Cushing syndrome (Table 17.4) [50–52]. Undetectable serum ACTH indicates an adrenocortical source of excessive cortisol causing suppression of ACTH secretion by the anterior pituitary. It can be particularly challenging to distinguish between anterior pituitary and ectopic sources of ACTH causing Cushing syndrome. Corticotroph adenomas, especially intrasellar ones, usually retain a partial capacity for negative feedback; the “high dose” dexamethasone suppression test, administered as eight 2-mg doses every 6 hours, can be expected to reduce cortisol levels by at least 50%. As this cause underlies the original description of the disorder by Harvey Cushing in 1912, pituitary-driven glucocorticoid excess is called Cushing disease. Extrapituitary tumors secreting ectopic ACTH less commonly display this degree of negative feedback such that ACTH levels and consequently cortisol levels are likely to be higher. Although these tumors may be relatively indolent carcinoids, more commonly they are aggressive carcinomas of the bronchus characterized by a rapid onset of symptoms within a few months, marked hypokalemia and weight loss. The high level of POMC gene expression and dysregulated post-translational processing also leads to significant melanocyte stimulating hormone (MSH) secretion resulting in skin pigmentation. Venous sampling of the bilateral inferior petrosal sinuses can distinguish between ectopic or anterior pituitary sources of excessive ACTH. For pituitary sources, a gradient in ACTH levels of at least 2:1 (or 3:1 after the injection of 100μg corticotropin releasing hormone [CRH] intravenously) should be found between central and peripheral samples. Although it can be very useful, bilateral inferior petrosal sinus sampling (BIPSS) also carries the risk of thrombosis in approximately 1% of cases, meaning that its use by endocrinologists is not always considered warranted. This is especially true where preceding biochemical tests have been conclusive and MRI shows clear radiologic evidence for a tumor. Where biochemistry is supportive of a pituitary source of ACTH excess, but the MRI is equivocal, even after gadolinium enhancement, BIPSS can help to lateralize a corticotroph adenoma by detecting a clear gradient between the right and left sinuses or vice versa. Sources of ectopic ACTH, most commonly in the chest, can be imaged with fine cut computerized tomography (CT), MRI or by using isotope-labeled scintigraphy to detect somatostatin receptors present on approximately two-thirds of ectopic ACTH-secreting carcinoid tumors. Autonomous cortisol-secreting tumors of the adrenal cortex can be imaged satisfactorily by CT or MRI when it can be possible to make assessment of functionality according to lipid content; features of high lipid content implying a tumor with active steroidogenesis.
Wherever possible, curative approaches are undertaken to normalize cortisol secretion as Cushing syndrome carries a high morbidity and markedly increases mortality from cardiovascular causes [50–52,58]. For adrenocortical sources, unilateral adrena-lectomy is performed. For ectopic ACTH-secreting tumors, excision of carcinoids is potentially curative, whereas the natural history of carcinoma of the bronchus with ectopic hormone secretion usually makes palliative care more appropriate. Medical therapy to treat corticotroph adenomas remains disappointing. Some excitement was gained for high dose peroxisome prolifer-ator-activated receptor γ (PPAR-β) agonists but larger trials have been disappointing. Pituitary tumors are treated by either trans-sphenoidal surgery or radiotherapy when the indications and complications are largely the same as for those described in the section on acromegaly. The fact that ACTH-secreting adenomas are commonly microadenomas can increase the chances of curative surgery, although this remains heavily operator-dependent. If surgery is delayed, suppressants of adrenocortical steroidogenesis such as ketoconazole or metyrapone can be used temporarily where patients are highly symptomatic [50–52]. In extremis, bilateral adrenalectomy provides a rapid resolution of excessive cortisol secretion, although the total loss of negative feedback to a corticotroph adenoma can result in dangerous pituitary tumor growth that becomes refractory to further treatment, a scenario called Nelson syndrome. In patients where ACTH levels continue to rise, concomitant radiotherapy to the anterior pituitary can help reduce the risk of this problem arising.
Outcome of Cushing syndrome and disturbance to glucose tolerance
After successful surgery, the patient is reliant on external glucocorticoid administered as oral hydrocortisone, but a return of cortisol secretion from the adrenal gland(s) can be anticipated over ensuing weeks. Commonly, a physiologic return of diurnal rhythm is never achieved postoperatively [59]. For some, the extensiveness of anterior pituitary surgery, or the long-term suppression either of normal corticotrophs (pituitary or ectopic ACTH secreting tumors) or normal adrenocortical cells (cortisol-secreting tumors) causes a permanent state of hypocortisolism because of underactivity of remaining tissue. This can require continued hydrocortisone administration as for patients with Addison disease, except that mineralocorticoid replacement should not be required.
With successful treatment of Cushing syndrome (Figure 17.7), glucose intolerance may resolve. In such instances, when the patient might be entirely dependent on replacement doses of hydrocortisone, care needs to be taken not to cause hypoglycemia by continued antidiabetes medication. By contrast, persisting abnormalities are relatively common, most likely associated with persistent visceral obesity and metabolic syndrome [59]. On close analysis in one case series, there was a marked persistence of visceral obesity and glucose intolerance in approximately 60% of patients who fulfilled criteria for remission of Cushing syndrome [60].
Effects of diabetes on hypothalamic–anterior pituitary–adrenal cortex axis
Evidence of overactivity of the hypothalamic–pituitary–adrenal (HPA) axis has been reported in children, adolescents and adults with diabetes, including moderate increases in urinary free cortisol excretion, plasma cortisol and ACTH concentrations [61–64]. These abnormalities have been related to the duration of diabetes [62,63] and to the presence of diabetic neuropathy [64]. Serum cortisol levels fall as metabolic control improves [65].
Pheochromocytoma
Etiology, incidence and clinical features of pheochromocytoma
Pheochromocytomas are catecholamine-secreting tumors arising from the chromaffin cells of the adrenal medulla (Figure 17.9) [66,67]. Approximately 10% lie in extra-adrenal sites (paragangliomas) along the sympathetic chain in para-aortic or chest regions and approximately 10% are bilateral. Although most pheochromocytomas are sporadic, advances in molecular genetics have demonstrated that a hereditary basis underlies approximately one-fifth to one-quarter of diagnoses, not just in well-known syndromes, such as MEN2, von Recklinghausen neurofibromatosis and von Hippel-Lindau disease [66]. Such advances have also associated particular genetic defects to tumor type. For instance, succinate dehydrogenase subunit B (SDHB) and succinate dehydrogenase subunit D (SDHD) mutations are more frequent in extra-adrenal tumors [66]. Mutations in SDHB are more commonly associated with a malignant phenotype [68]. The risk of malignancy also increases with tumor size [68].
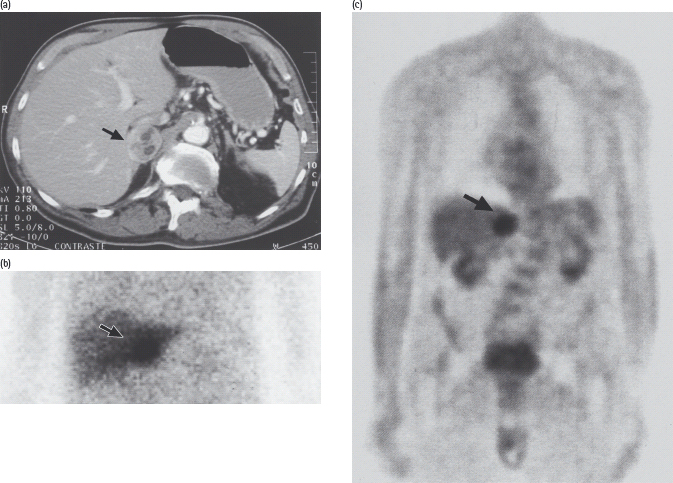
Figure 17.9 Pheochromocytoma in a 75-year-old man with recent-onset type 2 diabetes treated with a sulfonylurea, who was admitted on an emergency basis with severe chest pain and hypertension (blood pressure 240/130 mmHg). Blood glucose on admission was 27 mmol/L. Urinary catecholamine excretion was greatly increased, and a pheochromocytoma of the right adrenal was demonstrated by: (a) computed tomography; (b) scanning with 131I-metaiodobenzylguanidine, which is taken up by catecholamine-synthesizing tissues; and (c) positron emission tomography with 18F-fluorodeoxyglucose. After laparoscopic removal of the tumor, diabetes and hypertension both resolved.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


