INTRODUCTION
SUMMARY
Hemoglobinopathies are the most common inherited red cell disorders worldwide. Among these disorders, sickle cell syndromes and thalassemias constitute a major public health problem. A glutamic acid to valine substitution at the sixth amino acid of the β-globin chain of human hemoglobin (HbA) results in formation of sickle hemoglobin (HbS). Sickle cell disease results from homozygosity for this mutation, or from a compound heterozygosity for sickle hemoglobin and β-thalassemia or another β-globin variant such as HbC, HbD, HbE, or HbOArab. The sickle mutation renders the hemoglobin molecule insoluble upon deoxygenation; thus red blood cells containing deoxy HbS polymer are rigid and have impaired rheologic properties. The downstream effects of the sickling process include: membrane changes leading to potassium loss and cellular dehydration, interaction of sickle hemoglobin with microvascular endothelium, neutrophils, and monocytes, hemolysis, nitric oxide depletion, release of inflammatory proteins and activation of coagulation. These processes lead to a hemolytic anemia, an inflammatory state, painful vasoocclusive episodes, and damage to multiple organ systems with a resultant shortened life expectancy. There is considerable heterogeneity in the severity of the disease; the best known modifier of the disease is an elevated level of fetal hemoglobin (HbF), which exerts a potent antisickling effect. Concomitant α-thalassemia is also a modifier, which leads to a decrease in hemolysis. There is an interest in nonglobin genetic modifiers of sickle cell disease. Over the past 3 decades, advances in supportive care and implementation of disease-modifying therapies, such as anti–γ to β-globin switching therapies, which result in increased HbF and less HbS synthesis, and have led to an increase in life expectancy. Hydroxyurea has emerged as an effective disease-modifying agent that has been approved by the FDA for use in adults with sickle cell disease. Although its main mechanism of action is to enhance HbF production, other effects such as a decrease in neutrophils, platelets, and decreased expression of adhesion molecules contribute to its efficacy. Novel antiswitching agents, most notably, DNA methyltransferase 1 inhibitors (5′-azacytidine and decitabine) and histone deacetylase inhibitors (butyrate derivatives and others) are now in clinical trials. Evolving therapies include antiadhesive therapies to prevent interaction of sickle cells with microvascular endothelium, antiinflammatory approaches, and modulation of hemoglobin–oxygen affinity to prevent sickling. To date, the only curative therapy remains allogeneic hematopoietic stem cell transplantation.
Sickle trait, the heterozygous state for sickle hemoglobin, affects approximately 8 percent of Americans of African descent, and with rare exceptions is asymptomatic. HbC is associated with target cells and spherocytes in the blood film and splenomegaly. HbD disease is essentially asymptomatic. HbE is very common in Southeast Asia, and because of large population movements from this area, it has become a prevalent hemoglobinopathy in other regions of the world. HbE is a thalassemic variant and its coinheritance with β0-thalassemia mutations can result in severe transfusion-dependent thalassemia major. Unstable hemoglobin variants appear as rare, sporadic cases and are characterized by a Heinz body hemolytic anemia. Variants that alter the oxygen affinity of the Hb molecule lead to erythrocytosis (high oxygen affinity variants) or anemia (low oxygen affinity variants) and are rare causes of these syndromes.
Acronyms and Abbreviations
ACS, acute chest syndrome; ADMA, asymmetric dimethylarginine; AHSCT, allogeneic hematopoietic stem cell transplantation; BMP, bone morphogenic protein; 2,3-BPG, 2,3-bisphosphoglycerate; CO2, carbon dioxide; CSSCD, Cooperative Study of Sickle Cell Disease; eNOS, endothelial nitric oxide synthase; Hb, hemoglobin; HbAS, sickle cell trait; HbF, fetal hemoglobin; HbS, sickle hemoglobin; HbSC, sickle cell–HbC disease; HIF, hypoxia-inducible factor; HLA human leukocyte antigen; HPLC, high-performance liquid chromatography; IL, interleukin; iNKT cells, invariant natural killer T cells; K+, potassium; LDH, lactate dehydrogenase; MCHC, mean cell hemoglobin concentration; MCV, mean corpuscular volume; MPs, microparticles; MRI, magnetic resonance imaging; NO, nitric oxide; NT-pro-BNP, N-terminal pro–brain natriuretic peptide; O2, oxygen; P50, point at which hemoglobin is one-half saturated with oxygen; PCV7, pneumococcal polyvalent conjugate 7; PH, pulmonary hypertension; PIGF, placenta growth factor; PO2, partial pressure of oxygen; R state, relaxed oxy; SCD, sickle cell disease; SCT, stem cell transplantation; sPLA2, secretory phospholipase A2; STOP, Stroke Prevention Trial in Sickle Cell Disease; T state, tense, deoxy; TCD, transcranial Doppler; TF, tissue factor; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha; UDP, uridine diphosphate; UGT1A1, UDP glucuronosyltransferase 1 family; VOE, vasoocclusive episode; VTE, venous thromboembolism.
THE STRUCTURE AND FUNCTION OF NORMAL HEMOGLOBIN
The red protein hemoglobin (Hb) serves to transport oxygen from the lungs to the tissues and carbon dioxide (CO2) from the tissues to the lungs. Hb also binds the physiologically important nitric oxide (NO) molecule. The protein has evolved to perform its gas transport functions in a highly efficient manner. The oxygen affinity of Hb permits nearly complete saturation with oxygen in the lungs, as well as efficient oxygen unloading in the tissues because of its sigmoid oxygen dissociation curve. This curve results from the fact that Hb is a four-subunit, allosteric molecule; its conformation, and hence the oxygen affinity, changes as each successive molecule of oxygen is bound. Hb also plays an important role in acid–base balance: deoxyhemoglobin binds protons and oxyhemoglobin releases protons. Regulation of the oxygen dissociation curve to meet the needs of the body is remarkable. Hypoxic tissues become acidotic acutely, and the protons released produce a shift in the oxygen dissociation curve that enables more oxygen to be delivered to the tissue. However, longer-term acidosis or alkalosis (as occurs at high altitudes) is counteracted by modulation of red cell 2,3-bisphosphoglycerate (2,3-BPG), serving to decrease hemoglobin–oxygen affinity (Chap. 47).
Normal mammalian Hbs contain two pairs of related polypeptide chains: one chain of each pair is α or α-like and the other is non-α (β, γ, or δ). The α-chains of all human Hbs encountered after early embryogenesis are the same. The non-α chains include the β-chain of normal adult Hb (α2β2), the γ-chain of fetal Hb (α2γ2), and the δ-chain of the minor adult Hb (HbA2 [α2δ2]), which accounts for 2.5 percent of the Hb of normal adults. Chapter 48 discusses the regulation of production of the globin chains.
Certain residues in the amino acid sequence of each polypeptide chain appear to be critical to stability and function. Such residues are usually the same (invariant) in α or β chains. The NH2-terminal valines of the β chains are important in 2,3-BPG interactions. The C-terminal residues are important in the salt bridges that characterize the unliganded molecules. Areas of contact between chains and between heme and globin tend to contain invariant residues. The non-α (β, γ, δ, or ε) chains are all 146 amino acids in length. The γ-chain of fetal Hb (HbF) differs from the β-chain by 39 residues. The γ genes are duplicated: one codes for glycine (Gγ) and the other for alanine (Aγ)7 at residue 136, giving rise to two kinds of γ chains. In addition, a common polymorphism, the substitution of threonine for isoleucine, is frequently found at residue 75 of the Aγ-chain.
Approximately 75 percent of the amino acids in α and β chains are in a helical arrangement. All Hbs studied have a similar helical content (Fig. 49–1A). Eight helical areas, lettered A to H, occur in the β chains. Hb nomenclature specifies that amino acids within helices are designated by the amino acid number and the helix letter, whereas amino acids between helices bear the number of the amino acid and the letters of the two helices. Thus, residue EF3 is the third residue of the segment connecting the E and F helices, whereas residue F8 is the eighth residue of the F helix. Alignment according to helical designation makes homology evident: Residue F8 is the proximal heme linked histidine, and the histidine on the distal side of the heme is E7.
Figure 49–1.
A. Representation of the structure of β chains. Arrows indicate sites of substitutions in a number of unstable hemoglobins. B. The hemoglobin molecule, as deduced from x-ray diffraction studies, shown from above. The molecule is composed of four subunits: two identical α chains (light blocks) and two identical β chains (dark blocks). 2,3-BPG binds to the two β chains in the deoxyhemoglobin molecule. C. Schematic of rotation of α2β2 dimer relative to α1β1 in quaternary structure change from deoxyhemoglobin (solid lines) to carboxyhemoglobin (dashed lines). (Modified from Baldwin J, Chothia C: Haemoglobin: The structural changes related to ligand binding and its allosteric mechanism. J Mol Biol 129(2):175–220, 1979.)
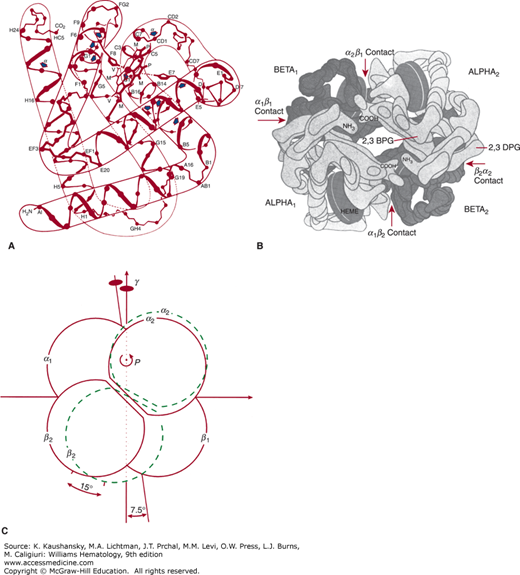
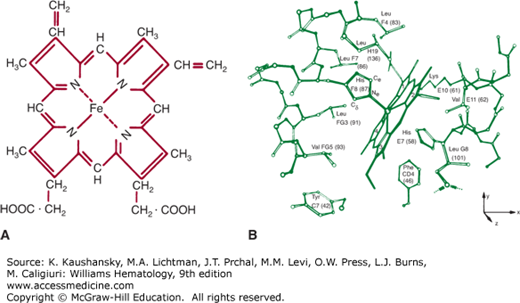
Figure 49–1B show the tertiary structure of the α and β chains. The prosthetic group of Hb is heme (ferroprotoporphyrin IX); Fig. 49–2A shows its structure. The heme group is located in a crevice between the E and F helices in each chain (Fig. 49–2B). The highly polar propionate side chains of the heme are on the surface of the molecule and are ionized at physiologic pH. The rest of the heme is inside the molecule, surrounded by nonpolar residues except for two histidines. The iron atom is linked by a coordinate bond to the imidazole nitrogen (N) of histidine F8. The E7 distal histidine, on the other side of the heme plane, is not bonded to the iron atom, but is very close to the ligand-binding site.
Figure 49–2.
A. Structure of heme (ferroprotoporphyrin IX). B. Heme group and its environment in the unliganded α-chain. Only selected side chains are shown; the heme 4-propionate is omitted. (Reproduced from Gelin BR, Lee AW, Karplus M: Hemoglobin tertiary structural change on ligand binding. J Mol Biol 25;171(4):489–559, 1983.)
The sigmoid oxygen dissociation curve is a function of the change of the conformation of the molecule from the liganded to the unliganded state (Table 49–1). In the deoxy state, the Hb tetramer is held together by intersubunit salt bonds (Fig. 49–3) and intersubunit hydrophobic contacts (see Fig. 49–1B), in addition to a certain number of hydrogen bonds. In deoxyhemoglobin, 2,3-BPG is situated in the central cavity between the two β chains (see Fig. 49–1B). The change in conformation of the Hb molecule is brought about by a complex, coordinated series of changes in the structure of the molecule as heme binds oxygen. The oxygen dissociation curve can be linearized by a transformation known as the Hill plot:
where K is an empiric overall constant without physicochemical basis. The slope n is taken as a convenient measure of cooperativity. Values of n in noninteracting Hbs that exhibit hyperbolic, not sigmoid, oxygen dissociation curves (e.g., myoglobin) are approximately 1. In a normal tetrameric Hb with four oxygen-reactive sites, the maximum value for n is 4.0; however, n values of 2.7 to 3.0 are found in normal Hb.
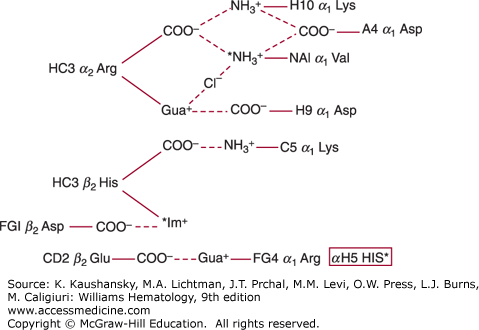
Figure 49–3.
Salt bridges in deoxyhemoglobin (* = ionizable group less protonated at pH 9.0 than at pH 7.0). These groups account for 60% of the alkaline Bohr effect. The remainder is due to αH5 His. (Data from Perutz MF, Wilkinson AJ, Paoli M, et al: The stereochemical mechanism of the cooperative effects in hemoglobin revisited, Annu Rev Biophys Biomol Struct 1998;27:1-34.)
The point at which the Hb is one-half saturated with oxygen (P50) is the usual measurement of oxygen affinity. It depends upon pH (the Bohr effect), temperature, and 2,3-BPG concentration. In common practice, P50 is standardized at 37°C and pH 7.20. P50 of freshly drawn blood is approximately 26.7 torr under standard conditions, but the partial pressure of oxygen (PO2) of Hb from which 2,3-BPG has been removed is only approximately 13 torr. Although fetal and newborn red cells have 2,3-BPG levels similar to those of adults, their oxygen dissociation curve is left shifted (increased oxygen affinity) with a P50 of approximately 23 torr because HbF does not react as strongly with 2,3-BPG as does HbA.
NOMENCLATURE OF ABNORMAL HEMOGLOBINS
Following the molecular characterization of HbS by Ingram and colleagues in 1956, there has been a rapid and exponential increase in the number of variant or “abnormal” Hbs.1 This number now exceeds 1000. A detailed description of variant Hbs, their chemical and functional properties, and population distribution can be found on the Globin Gene Server website (http://globin.cse.psu.edu/). Initially, newly described variants were designated by letters of the alphabet (e.g., HbC, HbD, HbE, HbJ). When the letters of the alphabet were exhausted, the practice of naming the variant Hbs after the geographic location where they were first described was adapted (e.g., HbKoln, HbZurich). Variants with electrophoretic or functional properties similar to previously described abnormal Hbs were designated with the letter and the geographic location (e.g., HbDPunjab, HbESaskatoon, HbMHyde Park). Some alphabetic designations were also used to indicate electrophoretic properties of certain variants; for example, there are a number of HbDs (DPunjab, DIran, DIbadan). All of these variants share the electrophoretic properties of HbS-like mobility on alkaline (cellulose acetate) electrophoresis, whereas they move with HbA at acidic pH (citrate agar electrophoresis). Similarly, HbEs have HbC-like mobility on alkaline electrophoresis and move with HbA on citrate agar electrophoresis.
The vast majority of Hb variants arise as a result of single nucleotide mutations, leading to an amino acid change in either α-, β-, δ-, or γ-globin subunits of the Hb tetramer resulting in variants of HbA (α or β), HbA2 (δ), or HbF (γ). Other mechanisms include small deletions or insertions, elongated chains, and fusions (for a detailed description of Hb variants and associated clinical syndromes, see “Other Abnormal Hemoglobins” below.
The coinheritance of HbS with some other variant Hbs or β-thalassemia mutations results in a number of sickling syndromes. In the United States, the most common sickling disorder is homozygous HbS (HbSS, sickle cell anemia), which is now commonly referred to as sickle cell disease (SCD). This is followed by sickle cell-HbC disease (HbSC), sickle cell–β+-thalassemia (HbS–β+-thalassemia), and sickle cell–β0-thalassemia (HbS–β0-thalassemia). Other rarer forms include HbSDPunjab, HbSOArab, HbSLepore, and HbSE diseases. Coinheritance of a large number of β-chain variants with HbS does not result in a symptomatic sickling disorder; rather, they are clinically and hematologically indistinguishable from sickle cell trait (HbAS).
HbC is found in 17 to 28 percent of West Africans, particularly east of the Niger River in the vicinity of North Ghana. The selective factors that account for this high prevalence are unknown at present, but HbC probably confers some resistance to infection with malaria. The prevalence of HbC among Americans of African descent is 2 to 3 percent. Sporadic cases also have been reported in other populations, including Italians and Afrikaners.
HbDPunjab, which is now recognized to be identical with HbDLos Angeles because both have the structure α2β2121 Glu→Gln, also interacts with HbS in forming aggregates in the deoxy conformation. HbD has been found in many parts of the world, including Africa, northern Europe, and India.
HbE is so prevalent that it may be the most common abnormal Hb or second in prevalence only to HbS. HbE is found principally in Burma, Thailand, Laos, Cambodia, Malaysia, and Indonesia. In some areas, HbE is found with a carrier rate of 30 percent. On the other hand, it is not prevalent among Chinese. Studies of restriction length polymorphisms in the β-globin cluster indicate the HbE mutation has arisen several times independently. It, too, probably confers some resistance to infection with malaria.
SICKLE CELL DISEASE
The first case of SCD, reported in 1910, was that of a dental student from Grenada, Walter Clement-Noel, studying in Chicago. Dr. James Herrick and his intern, Dr. Ernest Irons, were in charge of Mr. Noel’s care between 1904 and 1907, during which time he had several bouts of fever and cough and a history of leg ulcers, jaundice, and exercise intolerance. Herrick and Irons made astute clinical observations and prepared blood films and photomicrographs of nucleated red blood cells and of red cells having a “slender sickle shape” (Fig. 49–4).2 During the next decade, two more cases of this unusual anemia were reported. In 1915, Cook and Meyer raised the question of a genetic basis for the disorder based on the family history of the third reported case. In 1917, Victor Emmel used in vitro culture to show that sickle red cells represented a physical alteration of morphologically normally appearing red cells and were not released from the marrow as sickle cells.3 He also demonstrated that morphologically normal red cells of the father of a patient became sickle shaped after in vitro culture. Vernon Mason, who reported the fourth case in 1922, coined the term sickle cell anemia after observing the similarities between all the cases reported up to that time. In 1923, Sydenstricker and Huck noted “latent-sicklers” among relatives of the diagnosed patients, confirming and expanding on Emmel’s finding. In 1927, Hahn and Gillespie showed that sickling was related to low oxygen tension and low pH. In 1933, Diggs distinguished the difference of symptomatic cases called sickle cell anemia, from asymptomatic cases that were termed sickle cell trait, and he found that approximately 8 percent of Americans of African descent had the sickle cell trait.4
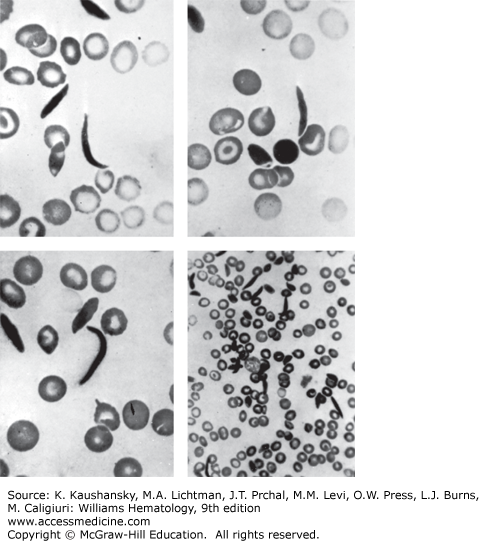
Irving Sherman, while a medical student at Johns Hopkins, showed that sickled red cells were birefringent under a polarizing microscope and that this finding was reversible with oxygenation of the cells. This observation ultimately led Linus Pauling to study sickle Hb after being advised of this property of sickle cell by William Castle, a noted research hematologist. Indeed, in 1949, Pauling and his colleagues demonstrated electrophoretic differences between Hbs from normal, sickle cell trait, and sickle cell anemia subjects and hypothesized that there must be chemical differences, thus establishing sickle cell anemia as the first molecular disease described. In the late 1950s, Hunt and Ingram sequenced the globin peptide and linked the abnormality to a change in the amino acid composition of the β-globin chain (replacement of glutamic acid by valine at residue 6). In 1977, Marotta and coworkers showed that the corresponding change in codon 6 of the β-globin gene was GAG→GTG. The discovery of a variant fragment in HbS versus HbA during restriction endonuclease mapping of amniotic fluid cells by Y. W. Kan paved the way for antenatal diagnosis of SCD and opened the way for modern genetics using recombinant DNA technology.5
The history of sickle cell anemia serves as an inspiring reminder of the power of clinical and laboratory observations, and in an era of mechanistic basic science research, serves to highlight the importance of bedside to bench and bench to bedside research integration.6,7,8,9
The observation that sickle cell trait may have a survival advantage against some environmental factors was first suggested by Dr. Alan Raper in East Africa in 1949. Drs. Mackey and Vivarelli suggested that the environmental influence might be malaria. It was subsequently noted that blood from sickle cell trait persons contained less malarial parasites and that the sickle trait conferred some protection against malaria in early childhood. Data suggest that sickle trait is maximally protective against severe malaria as opposed to asymptomatic parasitemia or mild disease.10 The mechanism of such a protection has been the matter of much debate. Plausible mechanisms include selective sickling of parasitized red blood cells, resulting in more effective removal by the monocyte-macrophage system, and inhibitory effect on parasite growth by increased red cell potassium loss, decreased red cell pH, and increased endothelial adherence of parasitized sickle red cells.
Thus, the prevalence of sickle cell anemia closely mirrors the worldwide distribution of falciparum malaria; however, as a result of migration of peoples to the industrialized Western countries, SCD has become more prevalent in areas where malaria is not endemic.
The World Health Organization estimated in 2006 that 5 percent of the world population carries a gene for a hemoglobinopathy. Sickle cell anemia is highly prevalent in sub-Saharan and equatorial Africa with lesser but significant prevalence in the Middle East, India, and the Mediterranean region. Incidence of SCD in sub-Saharan African countries ranges between 1 and 2 percent, which translates into approximately 500,000 cases per year. In the Jamaican cohort study, newborn screening in 100,000 consecutive vaginal deliveries resulted in the finding of sickle cell trait in 10 percent of newborns.11
In the United States, the Centers for Disease Control and Prevention estimates that sickle cell anemia is present in 1 in 500 livebirths among Americans of African descent; 1 in 12 American of African descent have the trait, and approximately 100,000 Americans largely of African descent live with the disease. In Americans of Hispanic descent, the rate of SCD is 1 in 36,000 livebirths. Accurate population statistics of SCD are difficult to obtain in the United States because of a lack of standardized data collection and central reporting.12
As of 2002, in the United States, more than one billion dollars are spent per year on hospitalizations for SCD.13 Data from a single state Medicaid program estimated a lifetime cost of care of $500,000 per patient with SCD. In this patient population, cost increased with increasing age, including cost of non-SCD health issues. The majority of the costs were for inpatient healthcare utilization.14
Previously, speculation existed as to whether the sickle mutation arose once and gained worldwide distribution or whether the mutation had arisen independently in different regions of the world. The nonrandom association of restriction endonuclease polymorphisms in the β-globin cluster define the β-globin haplotype. The β-globin gene cluster yields five distinct haplotypes associated with sickle cell mutations (Chap. 9).15,16,17 Four of the five patterns occur in Africa and are designated as the Senegal, Benin, Bantu, and Cameroon haplotypes, whereas the fifth arose on the Indian subcontinent.18 These findings indicate that the sickle mutation arose independently at five different times.
The sine qua non of sickle cell anemia is a Glu→Val substitution in the sixth amino acid of the β-globin gene. However, the pathophysiologic processes that result in the clinical phenotype extend beyond the red cell (Fig. 49–5). There is marked clinical heterogeneity from one patient to another and in the same patient over time. The heterogeneity for the same genotypic abnormality therefore implies that a multitude of other factors must contribute to the pathology of sickle cell anemia. The pathology is now far removed from the simplistic theory of hypoxia-induced microvascular occlusion. Sickle cell anemia is a chronic inflammatory state punctuated by acute increase in inflammation wherein the endothelium, neutrophils and monocytes, platelets, coagulation pathways, several plasma proteins, adhesion molecules, and derangements in NO metabolism interact in concert with the abnormality in Hb polymerization described several decades ago (Fig. 49–6). Abnormal adenosine signaling and activation of invariant natural killer T (iNKT) cells have been implicated in disease pathophysiology. Added to that are the complex differences in tissue-specific vascular beds and differences in various parts of the vasculature in the same organ. Also, variation in several genes other than the β-globin gene that modify the milieu in which organ damage occurs may play a role.
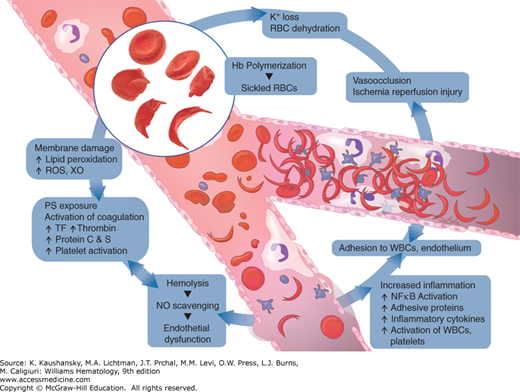
Figure 49–6.
Electron micrograph of negatively stained fiber of HgS and the structure deduced by three-dimensional image reconstruction. The reconstructed fiber is presented as ball models, with each ball representing a HgS tetramer. The models are presented as the outer sheath (left), the inner core (center), and a combination of both inner and outer filaments (right). (Reproduced with permission from the University of Texas Medical Branch.)

The pathophysiology of sickle cell anemia is described in separate sections; however, because no single, dominant pathway explains the multitude of manifestations, no single therapeutic modality serves to abrogate all of the pathology. Most experiments are in isolation in animal models or relatively simplistic experimental conditions with few in vivo studies in humans and thus do not replicate the complexity of this disorder.
Aggregation of deoxy HbS molecules into polymers occurs when aggregates reach a thermodynamically critical size. This process is termed homogenous nucleation, and the smallest aggregate formed that favors polymer growth is called the critical nucleus.19,20,21,22,23,24 Addition of subsequent deoxy HbS molecules to already formed polymers is termed heterogenous nucleation, which results in polymer branching. Polymer growth is, therefore, an exponential process wherein there is a delay time between presence of deoxy HbS molecules and polymer formation. This delay time is inversely proportional to the concentration of HbS molecules. Polymer formation alters the rheologic properties of the red cell.
The quaternary structure of oxy HbS cannot maintain axial and lateral hydrophobic contacts unlike that in the deoxygenated state, thus explaining the unsickling phenomenon upon reoxygenation.25,26,27,28 The sickling process that is initially reversible with oxygenation of deoxy HbS eventually leads to the formation of sickle-shaped red cells that fail to return to their normal discoid shape with oxygenation because of membrane damage imparted by repeated cycles of sickling and unsickling in the circulation. These cells are then termed irreversibly sickled cells. The rate and extent of polymerization is dependent on several factors, including intracellular Hb concentration, presence of Hbs other than HbS, blood oxygen saturation, pH, temperature, and 2,3-BPG levels.29 Microvascular occlusion by sickle red cells containing polymers is favored by prolonged transit times through the microcirculation, rapid deoxygenation and increased numbers of dense sickle red cells that contain polymers even at oxygen saturation levels found in the arterial circulation.29,30,31,32 Arguments against HbS polymerization as the major determinant of sickle cell pathophysiology include lack of clinically significant events despite constant sickling of red cells, the association of neutrophilia with vasoocclusive episodes (VOEs), and clinical features that imply macrovascular rather than microvascular perturbation, for example, large-vessel stroke.33
Membrane injury in HbSS red cells results in impaired cation homeostasis with decreased ability to maintain intracellular potassium concentrations. The calcium-activated potassium (K+) channel (Gardos channel), potassium-chloride cotransport channel, and a sickling-induced nonselective cation leak pathway have been implicated in sickle red cell dehydration. The net result is loss of intracellular potassium and water resulting in cellular dehydration.34,35,36,37,38,39 This change effectively increases the red cell Hb concentration, favoring sickling.
NO is a key component of the vascular endothelium that has vasodilatory, antiinflammatory, and antiplatelet properties.40 NO is a soluble gas synthesized from L-arginine by endothelial nitric oxide synthase (eNOS).41 Red cell L-arginase released as a consequence of sickle red cell hemolysis converts arginine to ornithine, thereby limiting L-arginine availability for NO synthesis. Decreased NO production because of elevated levels of endogenous nitric oxide synthase (NOS) inhibitors, especially asymmetric dimethylarginine (ADMA) and reduced L-arginine, have been documented in SCD especially during VOE.42,43,44,45,46 Reduced plasma arginine levels and elevated ADMA levels also result in NOS coupling causing production of reactive oxygen species rather than NO.47,48 Chronic hemolysis with release of plasma free Hb results in scavenging of NO with consequent endothelial dysfunction, which may favor sickle cell adherence.49,50
Seminal work by several groups showed that sickle red cells adhere to stimulated endothelium unlike their normal counterparts.51,52 Newly released red cells, called reticulocytes, express high levels of adhesion molecules, integrin α4β1, and CD36, and are more adherent than dense sickle red cells.53,54 Increased endothelial reticulocyte adhesion as compared to dense red cell adhesion is thought to be secondary to deformable red cells adhering to the endothelium behind which the dense red cells are trapped, leading to microvascular occlusion.29 Other molecules involved in sickle red cell-endothelium interactions include vascular cell adhesion molecule (VCAM)-1, integrin αVβ3, P-selectin, P-selectin glycoprotein ligand (PSGL)-1, E-selectin, Lutheran blood group antigen, and thrombospondin.55,56,57,58,59,60 The site of adhesion is purported to be the postcapillary venule at which site sickle red cells appear to interact with white cells adherent to the endothelium rather than engaging the endothelium directly.31
Neutrophilia is an adverse prognostic factor in sickle cell anemia. Because of their larger size, adherent leukocytes cause a greater decrease in vessel caliber than red cells. Diapedesis occurs in postcapillary venules, a site of vasoocclusion in sickle cell anemia.31,61,62,63 Neutrophil integrin αMβ2 microdomains capture sickle red cells causing vascular occlusion in sickle cell mouse models. Monocytes are also highly activated in sickle cell anemia, and they promote increased endothelial activation by increased production of tumor necrosis factor (TNF)-α and interleukin (IL)-1β.60 Expression of leukocyte adhesion molecules, L-selectin, and integrin αMβ2, are associated with a severe clinical phenotype.61,64
Sickle cell anemia is characterized by chronic leukocytosis, abnormal activation of neutrophils and monocytes, and an increase in several proinflammatory mediators including TNF-α, IL-6, and IL-1β. Several adhesion molecules are upregulated, including VCAM, selectins, integrins, the acute phase reactants C-reactive protein, secretory phospholipase A2 (sPLA2), and coagulation factors are activated.64–76 Placenta growth factor (PIGF) released from erythrocytes activates monocytes to produce inflammatory cytokines and upregulates endothelin-1 signaling via the endothelin B receptor. Endothelin-1 is a potent vasoconstrictor and upregulation is associated with adverse outcomes in SCD. Placental growth factor has independently been shown to be correlated with disease severity as well.77,78 Hemin has been demonstrated to activate PIGF in mice via the erythroid Kruppel-like factor; consequently, PIGF may play an important role in the pathophysiology of iron overload as well.79 It is an open question whether inflammation is caused by abnormally adhesive red cells to the vascular endothelium or whether inflammation causes abnormal red cell adhesiveness. It is likely both occur, given that red cell adhesiveness incites endothelial activity, and infection-induced inflammation precipitates clinically significant vascular events in patients.
The vascular beds in sickle cell anemia display changes akin to atherosclerotic vascular disease: large vessel intimal hyperplasia and smooth muscle proliferation.80,81 However, the characteristic lipid laden plaques of atherosclerotic vascular disease are not present.64
Akin to other disease states, such as myocardial infarction, resolution of vasoocclusion results in reperfusion injury characterized by increased oxygen free radical formation via activation of xanthine oxidase, generation of oxidant stress, lipid peroxidation, upregulation of cellular adhesion molecules, and nuclear factor-κB, a key player in the inflammatory process.64,82,83 iNKT cells propagate the inflammatory cascade in ischemia reperfusion injury and are increased and activated in patients with SCD. Agonists to adenosine 2A receptor (A2AR) on iNKT cells downregulate their activation and attenuate inflammation in mouse models of SCD.84
The initiator of coagulation, tissue factor (TF), is elevated in patients with sickle cell anemia.40,74,85,86,87 Microparticles (MPs) expressing TF derived from monocytes, macrophages, neutrophils and endothelial cells have been described in SCD.58,68,74,88 Conflicting results exist in the literature on the presence and contribution of TF bearing MPs. There is a lack of correlation between TF bearing MPs and procoagulant activity in SCD. Erythrocyte and platelet MPs are TF-negative and are the major component of MPs in SCD. Activation of the intrinsic pathway of coagulation by TF-negative, red cell, and platelet MPs through a phosphatidylserine-dependent mechanism appears to be the major contributor of MP-dependent coagulation activation in SCD. Perivascular TF interaction with plasma coagulation factors made possible by increased vascular permeability and phosphatidylserine exposure on the surface of red cells secondary to repeated cycles of sickling provide an impetus for the coagulation process.89 Heightened thrombin generation, platelet activation, and decreased protein C and S levels favor a procoagulant state.69,90,91 Increased plasma levels of D-dimers, thrombin–antithrombin complexes, prothrombin fragment 1.2, and plasmin–antiplasmin complexes are indicative of increased thrombin-mediated coagulation with subsequent fibrinolysis.92 Plasma from sickle cell patients contains increased ultralarge von Willebrand factor multimers as a result of increased endothelial cell secretion and impaired cleavage by ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif member 13).93
Cellular stress leads to the degradation of adenine nucleotides, resulting in the generation of adenosine. Adenosine homeostasis is maintained by two enzymes: adenosine kinase, which phosphorylates adenosine to adenosine monophosphate and adenosine deaminase, which converts adenosine to inosine. Adenosine signals through four different receptors that have differing functions. Signaling via the A2AR expressed on most leukocyte and platelets results in an antiinflammatory effect; however, signaling via the A2BR was shown to cause priapism in SCD mice via hypoxia-inducible factor (HIF)-1–mediated decrease of phosphodiesterase 5. Signaling via A2BR also leads to increased 2,3-BPG in red cells causing decreased oxygen binding affinity of Hb, which promotes sickling. Pegylated adenosine deaminase treatment of sickle mice resulted in decreased hemolysis and hypoxia reoxygenation injury.94,95
Inheritance of only one HbS allele is termed sickle cell trait (HbAS). An estimated 300 million people carry the trait worldwide.96 The percentage of HbA is always higher (~60 percent) than HbS (~40 percent) in sickle cell trait.
HbAS is considered a generally asymptomatic state with HbA in the cell preventing sickling except in the most unusual circumstances. HbAS cells sickle at O2 tension of approximately 15 torr.97
Plasma myeloperoxidase and red cell sickling have been reported to increase during exercise with fluid restriction in HbAS subjects.98 Plasma levels of VCAM-1 are higher in HbAS subjects and remain elevated following exercise compared to normal controls or HbAS with concomitant α-thalassemia, which is suggestive of subtle microcirculatory dysfunction in this population.99 Skeletal muscle capillary structures are different in HbAS subjects compared to controls. There is a 30-fold increased risk of sudden death in black army recruits with HbAS.100 Although controversial, in 2009 the National Collegiate Athletic Association recommended mandatory testing for HbAS for all its student athletes.101
Renal abnormalities are among the most common manifestations of HbAS. Anoxia, hyperosmolarity, and low pH of the renal medulla predisposes to sickling. Microscopic or gross hematuria from renal papillary necrosis is usually painless. Renal neoplasm or stones should be excluded in those with persistent gross hematuria. Isosthenuria may be seen in and may contribute to exercise induced rhabdomyolysis and sudden death.102 Renal medullary carcinoma is a rare but serious complication of HbAS. Risk of urinary tract infection is higher in females with HbAS, especially during pregnancy. End-stage renal disease occurs at an earlier age for HbAS patients with polycystic kidney disease and HbAS may contribute to erythropoietin resistance.103
Splenic infarction occurs under extreme environmental conditions in persons with HbAS; most resolve spontaneously.104,105 Caution and immediate intervention is also warranted in those HbAS individuals who develop traumatic hyphema.106 The risk of venous thromboembolism is increased twofold in HbAS subjects compared to those without the trait. The risk appears to be greater for pulmonary embolism than for deep vein thrombosis.101,105 HbAS patients do not have increased perioperative morbidity or mortality. The life span of patients with HbAS is normal.107
Sickle cell anemia is characterized by a laboratory profile of evidence of hemolytic anemia with increases in lactate dehydrogenase (LDH), indirect bilirubin, reticulocyte count, and a decrease in serum haptoglobin. Anemia is usually normochromic, normocytic with a steady-state Hb level between 5 and 11 g/dL.1,108 The red cell density is increased with a normal mean cell Hb concentration (MCHC).109 Serum erythropoietin level is decreased relative to the degree of anemia.110 Elevated neutrophil and platelet levels are observed even in asymptomatic patients reflective of persistent low-grade inflammation.111,112,113
Plasma tocopherol and zinc levels are low.114,115,116 Serum ferritin is increased, especially in iron overloaded patients. Elevated brain natriuretic peptide is seen in patients with pulmonary hypertension (PH) and congestive heart failure. Morphologically, classic sickle red cells are seen on blood film examination, and the marrow shows erythroid hyperplasia.
Sickle cell anemia can be accurately diagnosed with high-performance liquid chromatography (HPLC) and isoelectric focusing.117 Rapid methods, such as solubility testing and sickling of red cells using sodium metabisulfite, are less-reliable tests.118 Polymerase chain reaction is the method of choice for prenatal diagnosis.119 No HbA is found in patients with HbSS, HbSC, or HbSβ0 diseases. Varying amounts of HbA (depending on the severity of the β-thalassemia mutation) are found in HbS–β+-thalassemia subjects.
Mortality from SCD in the United States has declined since 1968, coinciding with the introduction of pneumococcal polyvalent conjugate 7 (PVC7) vaccine. Comparison of mortality rates between 1979 to 1998 and 1999 to 2009 showed a 61 percent decrease in infants, 67 percent in children ages 1 to 4 years, and 35 percent decrease in children ages 5 to 19 years. Transition from pediatric to adult medical care showed an increased mortality trend with similar rises in rates during the decades of comparison.120 Average life expectancy of patients with HbSS disease in the United States is 42 and 48 years for males and females, respectively.121 In Jamaica, the population has a median survival of 53 years and 58 years for men and women, respectively, with 44 percent of individuals born prior to 1943 still living as of 2009.122 As the sickle cell population ages, causes of death change from an infectious etiology to those related to end-organ damage, such as renal failure.
The reader is referred to the National Institutes of Health, National Heart, Lung and Blood Institute’s guidelines from 2002 for an extensive review on the topic; revised guidelines were released in the fall of 2014 at .123 General approaches to SCD management and pain management are described separately (Table 49–2).
| Pathophysiology/Complication | Therapeutic Interventions |
|---|---|
| Sickle hemoglobin (HbS) polymerization | Fetal hemoglobin (HbF) induction |
| Cellular dehydration | Gardos channel inhibition Potassium-chloride cotransport channel inhibition |
| Adhesion to endothelium | |
| Red blood cell | Antiselectin Antiintegrin |
| White blood cell | Antiselectin Intravenous immunoglobulin Hydroxyurea (HU) |
| Inflammation | Nuclear factor-κB inhibition Immunomodulatory drugs HU Statins |
| Nitric oxide (NO) scavenging | NO donor (NO, HU, tetrahydrobiopterin) Phosphodiesterase 5 inhibition Modulation of hemolysis |
| Coagulation | Tissue factor inhibition Antiplatelet therapy Anticoagulation |
| Hyposplenism/infection | Penicillin prophylaxis |
| Ischemia–reperfusion | Xanthine oxidase inhibition Myeloperoxidase inhibition |
| Iron overload | Chelation |
The typical course for a sickle cell patient is that of periods of relatively normal functioning despite chronic anemia and ongoing vasoocclusion, punctuated by periods of increased pain, and serial changes in various laboratory parameters that is termed “a sickle cell crisis.” Crises have typically been classified as VOEs, aplastic crises, sequestration crises, and hyperhemolytic crises.
Vasoocclusive Crises The hallmark of SCD is the VOE. It is the most common clinical manifestation but occurs with varying frequency in different individuals. It results from increasing vasoocclusion causing tissue hypoxia, which manifests as pain. Vasoocclusion may affect any tissue, but patients typically have pain in the chest, lower back, and extremities. Abdominal pain may mimic acute abdomen from other causes. Different patients display different patterns of painful sites during a VOE, but each patient’s recurrences usually mimic the same pattern of pain. Fever is often present, even in the absence of infection. Episodes may be precipitated by dehydration, infection, and cold weather although in about most cases no precipitating factor is found.124
Figure 49–7 illustrates the phases of VOEs.125 Crises requiring readmission within 1 week occur in approximately 20 percent of patients after hospital discharge.125
Figure 49–7.
A typical profile of the events that develop during the evolution of a severe sickle cell painful crisis in an adult in the absence of overt infection or other complications. Such events are usually treated in the hospital with an average stay of 9 to 11 days. Pain becomes most severe by day 3 of the crisis and starts decreasing by day 6 or 7. The Roman numerals refer to the phase of the crisis: I indicates prodromal phase; II, initial phase; III, established phase; and IV, resolving phase. Dots on the x-axis indicate the time when changes became apparent; and dots on the y-axis, the relative value of change compared with the steady state indicated by the horizontal dashed line. Arrows indicate the time when certain clinical signs and symptoms may become apparent. Values shown are those reported at least twice by different investigators; values that were anecdotal, unconfirmed, or that were not reported to occur on a specific day of the crisis are not shown. CPK, creatinine phosphokinase; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; HDW, hemoglobin distribution width; ISC, irreversibly sickled cells; LDH, lactate dehydrogenase; RBC DI, red cell deformability index; RDW, red cell distribution width; SAA, serum amyloid A. (Reproduced with permission from SK Ballas, K Gupta, P Adams-Graves: Sickle cell pain: A critical reappraisal. Blood 120(18):3647–3656, 2012.)
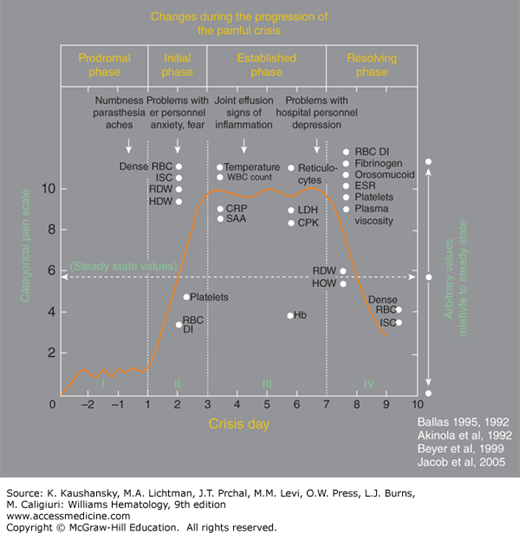
The characterization of crisis phases has implications for clinical research, especially in pain management, wherein interventions early in the course of a crisis could result in better outcomes for patients.
Aplastic Crises Aplastic crises in sickle cell anemia result when there is a marked reduction in red cell production in the face of ongoing hemolysis, causing an acute, severe drop in Hb level. The characteristic laboratory finding is a reticulocyte count less than 1 percent. The most common causative agent is parvovirus B19, which attaches to the P antigen receptor on erythroid progenitor cells, causing a temporary arrest in red cell production (Chap. 36). Recurrent aplastic crises by parvovirus B19 are rare because of the development of protective antibodies. Other rare complications associated with parvovirus B19 include acute splenic and/or hepatic sequestration, acute chest syndrome, marrow necrosis, and renal dysfunction. Patients usually recover within 2 weeks; however, those with severe symptomatic anemia need red cell transfusion. Siblings of SCD patients with parvovirus infections should be monitored closely for aplastic crisis given high secondary attack rates (>50 percent). Patients need to be isolated from pregnant individuals given increased risk of hydrops fetalis with parvovirus B19 infection.126
Sequestration Crises This type of crisis is characterized by sudden, massive pooling of red cells, typically in the spleen and less commonly in the liver.127 Splenic sequestration is typically seen in children (younger than 5 years of age) prior to autoinfarction of the spleen, but can be seen in adults with HbSC disease or HbS–β-thalassemia with persisting splenomegaly.128,129,130 A minor sequestration episode is usually accompanied by a Hb of more than 7 g/dL, and a major episode usually is one in which the Hb is less than 7 g/dL or the Hb has decreased by 3 g/dL from baseline.131
Acute splenic and hepatic sequestration crises can present with rapidly enlarging spleen or liver, pain, hypoxemia, and hypovolemic shock. Treatment consists of red cell transfusion. Transfusion carries the risk of hyperviscosity when the sequestration crisis resolves and the sequestered red cells are returned to the general circulation. Splenic sequestration crisis has a high rate of recurrence, especially in children. Splenectomy to prevent recurrence is debated in very young children. Some report chronic red cell exchange transfusion as a means of delaying splenectomy until the child is older while others did not see any benefit to this treatment. Patients younger than 2 years of age can be placed on chronic transfusion until they are older, at which time splenectomy should be considered. Splenectomy is recommended after the first episode of life-threatening splenic sequestration crisis or chronic hypersplenism. Partial splenectomy and emergency splenectomy during a crisis is not recommended. Parental education is important for early recognition of the problem so they can seek medical care promptly.126
Hyperhemolytic Crisis The term hyperhemolytic crisis is used to describe the occurrence of episodes of accelerated rates of hemolysis characterized by decreased blood Hb, increasing reticulocytes, and other markers of hemolysis (hyperbilirubinemia, increased LDH). Hyperhemolysis can occur during resolution of a VOE, at which time irreversibly sickled and dense red cells are rapidly destroyed, as well as from an acute or delayed hemolytic transfusion reactions.126,132
Patients with SCD have acute pain, chronic pain, or both. As a symptom, pain is often underrated in its intensity and undertreated by caregivers, especially inexperienced physicians. Patients are often perceived as drug-seekers or drug addicts, when in fact less than 10 percent of patients are addicted, a number comparable to other disease states. Unsatisfactory relief of pain drives patients to behaviors that appear to healthcare givers as signs of addiction—a state termed pseudoaddiction. A study comparing sickle cell anemia patients who use the emergency department frequently or infrequently found significant impairment in quality of life and increased markers of disease severity in those who use the emergency department frequently, dispelling the myth that frequent emergency department use indicates narcotic-addicted individuals when, in fact, they may have more severe disease.3,133,134,135,136,137 The landmark Pain in Sickle Cell Epidemiology Study revealed that adult SCD patients have pain at home approximately 55 percent of the time, which contrasts sharply to pain studies in children, who report at-home pain approximately 9 percent of the time.138,139
Acute pain is managed with opioids, nonsteroidal antiinflammatory drugs, acetaminophen, or a combination of these medications. Immediate pain assessment and frequent reassessment with appropriate application of medications until pain relief is obtained is important. For adults and children weighing more than 50 kg, morphine can be started at a dose of 0.1 to 0.15 mg/kg. The hydromorphone dose should be 0.015 to 0.020 mg/kg intravenously. These are recommended doses for opioid-naïve patients and are at the lower end of the dosing range.123,140,141 The use of meperidine has declined because of neurologic side effects, especially in patients with renal failure, who are at risk for the serotonin syndrome in conjunction with use of other medications.142,143,144 However, the use of morphine is not benign and concerns of increased association of acute chest syndrome, dysphoria, and neuroexcitatory side effects have been raised.125 Prior use of opioid therapy should be taken into consideration when deciding initial opioid doses as patients may be tolerant and require higher doses. Caution should be exercised with nonsteroidal antiinflammatory drugs and acetaminophen if there is renal or hepatic dysfunction. Patients with acute pain are better managed in a setting dedicated to sickle cell patients.145 A multidisciplinary approach is needed for pain management, especially if chronic pain is present.146,147 Opioid side effects should be anticipated and managed. Antidepressants, anticonvulsants, and clonidine can be used for neuropathic pain. Occasionally, severe, unrelenting pain may require red cell transfusion to decrease sickle Hb below 30 percent in the blood.148
There is a paucity of data regarding optimal management of pain in SCD. A randomized trial of optimizing patient controlled analgesia strategy was closed because of poor accrual.149 A trial looking at NO inhalation for treatment of VOE did not show improvement in pain.150
Acute Chest Syndrome The acute chest syndrome (ACS) is a constellation of signs and symptoms in patients with SCD that includes a new infiltrate on chest radiograph defined by alveolar consolidation but not atelectasis, chest pain, fever, tachypnea, wheezing, or cough, and hypoxia (Fig. 49–8).151 However, respiratory findings on clinical examination in the absence of radiographic findings should trigger high suspicion for ACS and warrants close monitoring. ACS is the leading cause of mortality in patients with SCD.121 Etiology varies depending on age, with viral and bacterial infections dominating in the pediatric age group and fat embolization resulting from marrow necrosis during VOE dominating in adults.152,153 Important pathogens include Chlamydia pneumoniae, Mycoplasma pneumoniae, Streptococcus pneumoniae, Staphylococcus aureus, parvovirus B19, respiratory syncytial virus, and influenza. Regardless of the triggering factor, the pathogenesis of ACS involves increased intrapulmonary sickling, intrapulmonary inflammation with increased microvascular permeability, and alveolar consolidation. ACS can rapidly evolve with bilateral infiltrates and consolidation leading to acute respiratory failure requiring intubation and ventilatory assistance.
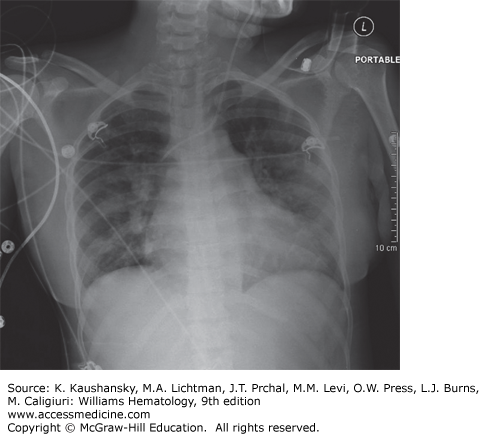
Independent risk factors for respiratory failure are age older than 20 years, platelet count less than 20 × 109/L, multilobar lung involvement, and a history of cardiac disease.152 Thrombocytopenia is an independent predictor of neurologic complications during hospitalization for ACS, which was seen in 22 percent of adult patients in the National Acute Chest Syndrome study.154
The treatment of ACS includes oxygenation, incentive spirometry, adequate pain control to avoid chest splinting, antimicrobial therapy that always covers atypical bacteria and influenza when indicated, avoidance of overhydration, use of bronchodilators, and red cell transfusion to decrease intrapulmonary sickling.152,155,156,157,158,159,160 The use of glucocorticoids may attenuate the course of ACS; however, its use is not well established and readmission rates for VOE after ACS resolution are increased.153 sPLA2 has been recognized as a predictor of ACS; however, a clinical trial investigating early transfusion based on sPLA2 elevation closed because of poor accrual. Hydroxyurea therapy should be offered to all patients with a history of ACS because it reduces the incidence by 50 percent in adults and 73 percent in children.161
Pulmonary Hypertension PH, defined by a resting mean pulmonary arterial pressure of 25 torr or higher on right-heart catheterization, is seen in 6 to 11 percent of SCD patients. An elevated tricuspid regurgitant velocity of 2.5 m/s has a positive predictive value of 25 percent for PH in SCD and is seen in one-third of these patients. PH, as defined by right-heart catheterization, elevated tricuspid regurgitant jet velocity of 2.5 m/s or higher, and a serum N-terminal pro–brain natriuretic peptide (NT-pro-BNP) level of 160 pg/mL or higher, confers an increased mortality risk.162
Abnormalities in NO metabolism, hemolysis, and inflammation contribute to the pathophysiology of PH.162 Parenchymal lung disease from repeated episodes of ACS and thromboembolism are other causal factors.
Clinical symptoms of PH include fatigue, dizziness, and dyspnea on exertion, chest pain, and syncope. These may be unrecognized as being related to PH, as PH is often undiagnosed in patients with SCD.
PH should be treated following guidelines set for the treatment of primary PH unrelated to SCD. Two trials looking at bosentan (endothelin receptor antagonist) in SCD patients closed because of sponsor withdrawal. A trial of sildenafil was halted early because of increased incidence of VOE. Patients who have venous thromboembolism in the setting of PH should be considered for indefinite anticoagulation. Hydroxyurea should be offered to all patients with any of the risk factors for increased mortality described above.162
Asthma, Abnormal Pulmonary Function Tests, and Airway Hyperreactivity Asthma is a common comorbidity with higher-than-average prevalence in patients with SCD and is associated with increased risk of ACS, VOE, stroke, and mortality. Airway hyperreactivity as evidenced by a positive bronchodilator response on pulmonary function testing, irrespective of baseline function, and in response to cold air or methacholine challenge, is seen in approximately two-thirds of SCD patients. Inflammation, hypoxemia, and increased oxidative stress associated with asthma may contribute to the vasculopathy of SCD.163
Pulmonary function tests collected as part of the Cooperative Study of Sickle Cell Disease (CSSCD) revealed abnormalities in 90 percent of the 310 patients, with the majority having restrictive lung disease.
Anemia in SCD results in an elevated cardiac output secondary to an increased stroke volume with minimal increase in heart rate.166,167 Clinical manifestations of a hyperdynamic circulation include a forceful precordial apical impulse, systolic and diastolic flow murmurs, and tachycardia that may increase during periods of increased hemodynamic stress. Diastolic left ventricular dysfunction may begin in early childhood and is an independent risk factor for death, with even greater risk of mortality in those having PH. Left ventricular hypertrophy is common and progressive with age; left ventricular dysfunction is a late event. Myocardial infarction is an underrecognized problem in SCD. Epicardial coronary artery disease is rare; microvascular ischemia is likely causative. Sudden cardiac death has been reported in 40 percent of patients in an autopsy series.168,169,170 Previously sudden cardiac death was ascribed to narcotic overdose; currently, it is thought to be secondary to cardiopulmonary causes in the majority of cases. QTc prolongation, atrial and ventricular arrhythmias, nonspecific ST-T wave changes are common in SCD patients. Patients presenting with chest pain should have a thorough evaluation to rule out cardiac disease. Cardiac magnetic resonance may be a good modality to image microvascular flow and quantitate cardiac iron overload.171,172 Blood pressure in patients with SCD is significantly lower than age-, sex-, and race-matched controls, partly secondary to anemia.173 Relative hypertension is associated with end-organ damage. Diuretics may be used, keeping in mind that SCD patients have obligate hyposthenuria and are prone to dehydration, which can precipitate a VOE.
Originally thought to be a small vessel disease, stroke in SCD is a macrovascular phenomenon with devastating consequences that affects approximately 11 percent of patients younger than 20 years of age.174,175 Risk is highest in the first decade of life followed by a second smaller peak after age 29 years. Ischemic stroke is most common in children and older adults, whereas hemorrhagic stroke predominates in the third decade of life.175 Recurrent stroke is most common in the first 2 years following the primary event.176 Silent infarcts, defined as an increased T2 signal abnormality on magnetic resonance imaging (MRI), begins in infancy and has a cumulative incidence of 37 percent by age 14 years. They occur in watershed areas of the brain, are not predicted by abnormal transcranial Doppler (TCD) velocity, and may progress despite chronic transfusion.177,178,179,180 There is evidence of neurocognitive decline in asymptomatic adults despite having normal brain imaging that is attributed to anemia and hypoxemia.154
Cerebral blood flow is significantly increased in SCD because of chronic anemia and hypoxemia, but does not increase further in response to increased hypoxic stress, thereby predisposing to ischemia.181,182 Stenosis of large vessels, especially of the circle of Willis, without the classic atherosclerotic plaque occurs in conjunction with a multitude of other factors, including chronic hemolysis, deranged NO metabolism and impaired vascular autoregulation, and can lead to stroke.182 Rare causes of cerebral vascular disease include fat embolization and venous sinus thrombosis. Moyamoya type fragile collaterals have been reported in more than one-fifth of patients with prior stroke, possibly leading to hemorrhagic stroke in later life.183,184,185,186,187,188
Risk factors for ischemic stroke include transient ischemic attack, recent or recurrent ACS, nocturnal hypoxemia, silent infarcts, hypertension, elevated lactic dehydrogenase, and leukocytosis, whereas anemia, neutrophilia, the use of glucocorticoids, and recent transfusion are independent risk factors for hemorrhagic stroke, especially in children.175,189,190,191,192,193,194,195 Sickle cell genotypes other than HbSS carry a lower risk, as do patients with HbS–α-thalassemia.175,196,197 The best predictor of stroke risk, however, is an increased blood flow velocity in major intracranial arteries on TCD ultrasonography.197 Blood flow velocities less than 170 cm/s are considered normal. Velocities between 170 and 200 cm/s are termed conditional, and velocities of greater than 200 cm/s are considered high and are associated with a 10-fold increase in ischemic stroke in children 2 to 16 years of age.
There is an increased frequency of stroke among siblings of patients with SCD than would be expected by chance alone, raising the possibility of other modifier genes contributing to stroke risk.183 The TNF (–308) G/A promoter polymorphism is associated with increased large-vessel stroke risk as is the IL-4–receptor gene 503 S/P variant, although it did not reach statistical significance. The clinical features of stroke in SCD encompass the classic findings of stroke in other disorders, including, but not limited to, hemiparesis, seizures, coma, paresthesias, headaches, and cranial nerve palsies. Neurocognitive deficits in IQ, memory, language, and executive function have been demonstrated.154,198
Imaging approaches for acute stroke are the same as those for non-SCD patients and includes MRI and magnetic resonance angiography.
Prevention of Primary Stroke Based on the results from the Stroke Prevention in Sickle Cell Disease (STOP) Study, it is recommended that asymptomatic children with HbSS disease older than two years of age should be screened for stroke risk using TCD.197 Those with high TCD velocities should be offered a chronic red cell transfusion program for primary stroke prevention. Repeat TCD screenings should be done every 3 to 12 months even in patients who have normal or conditional baseline velocities, because they can evolve into a higher-risk category. Despite obstacles to TCD screening, clinical practice changes based on the STOP study translated into declining stroke rates since 1991.199,200
Prevention of Secondary Stroke Patients with SCD who present with a stroke and are not on chronic transfusion should be placed on a transfusion program to prevent secondary strokes. Exchange transfusion may be preferable to periodic red cell transfusion, not only to avoid iron overload, but also to further reduce stroke risk. In a retrospective study, children who received periodic transfusion had a fivefold higher relative risk of a recurrent stroke compared to those on an exchange transfusion regimen.201 Despite chronic transfusions, patients may have a recurrent stroke, especially in patients with HbS greater than 30 percent.202 Hydroxyurea was shown to decrease high and conditional TCD velocities in more than 90 percent of patients studied.203 However, a randomized trial comparing transfusions with iron chelation to hydroxyurea with phlebotomy showed a 10 percent stroke rate in the hydroxyurea arm, thus establishing transfusion as the preferred preventive strategy.204
Anticoagulation therapy has not been studied in patients with SCD and, therefore, no recommendations can be made. Treatment guidelines for intracranial hemorrhage are as those for non-SCD–related intracranial hemorrhage; role of transfusion is less clear in SCD especially when cause of intracranial hemorrhage is unclear. Patients with moyamoya disease who have a particularly poor outcome may benefit from revascularization using encephaloduroarteriosyangiosis.205,206
Renal Failure Sickling of HbSS erythrocytes in the hypoxic, acidic, and hypertonic environment of the renal medulla, oxidative stress, increase in prostaglandins and endothelin-1 in the kidney, and abnormalities of the renin angiotensin system contribute to the pathophysiology of renal disease in SCD.207 The incidence of renal failure varies between 4 and 20 percent.208,209,210,211 Dehydration is the most common cause of acute renal failure in SCD. Isosthenuria is highly prevalent in SCD, may increase the risk of dehydration, and is irreversible.212 Glomerular hypertrophy, focal and segmental glomerular sclerosis, and hemosiderin deposition in proximal renal tubular epithelium have been described; however, no single lesion is pathognomonic of sickle cell nephropathy. Cystatin C is an accurate marker of glomerular filtration and therefore is preferable to serum creatinine in estimating renal function.213,214 Glomerular hyperfiltration, microalbuminuria, and macroalbuminuria occur sequentially in SCD patients starting in infancy and increasing in frequency with age.122,161,215 Incidence of microalbuminuria is greater than 60 percent in those over age 35 years.213 End-stage renal disease requiring dialysis carries a poor prognosis and is associated with a median survival of 4 years.216
Angiotensin-converting enzyme inhibitors decrease proteinuria and hyperfiltration in SCD; however, large-scale studies are needed to characterize the magnitude of the benefit. Treatment of renal disease follows principles used for non-SCD kidney pathology and includes effective blood pressure control, avoidance of nephrotoxic agents, and treatment of urinary tract infection. A relative decrease in serum erythropoietin levels, proportionate to the degree of anemia is observed; however, erythropoietin treatment, with its resultant increase in Hb may cause an increase in VOEs because of an increase in blood viscosity.213
Renal tubular acidosis type IV, secondary to decreased potassium and hydrogen ion in the distal tubule can cause disproportionate acidosis and hyperkalemia in patients with declining renal function.213
Hematuria is discussed in the section on sickle cell trait.
Priapism Priapism is prevalent in at least 35 percent of male patients with SCD with devastating psychological consequences; true prevalence may be higher as it is often underreported.217,218,219 The mean age of episodes is 15 years and two-thirds of patients have “stuttering priapism” a term used for episodes that last less than 3 hours.220 Derangements in NO metabolism and adenosine signaling are thought to be the major contributors to priapism in SCD.94 Greater than 95 percent of priapism is the “low-flow” type resulting from ischemia, is painful, and is a medical emergency.221
Aspiration of the corpus cavernosa followed by epinephrine injections, exchange transfusion, and α and β agonists have all been used, but data regarding efficacy are sparse. α-Agonists, etilefrine 50 mg, and ephedrine 15 to 30 mg per day, seem to reduce the incidence of stuttering priapism.222 Hormonal therapies, including antiandrogens and luteinizing hormone-releasing hormone, reduce nocturnal erections but are associated with loss of libido.221 Transfusion therapy has resulted in neurologic sequelae termed “the ASPEN syndrome” (Association of Sickle Cell Disease, Priapism, Exchange Transfusion) and is thought to be secondary to hyperviscosity; care, therefore, must be taken not to increase the hematocrit beyond 30 percent.223 In recalcitrant cases, a shunt is performed but results in permanent impotence.222 A penile prosthesis is used to ameliorate sexual dysfunction.
Nocturnal Enuresis Nocturnal enuresis is prevalent in 25 to 33 percent of the pediatric sickle cell population, which is higher compared to that of age-matched controls.224,225,226 It tends to decrease with age but is still prevalent in adults. Social and environmental factors, decreased functional bladder capacity, and decreased arousal during sleep appear to be contributing factors.
VOE is commonly manifested by marrow infarction causing musculoskeletal pain, swelling at involved sites, fever, and leukocytosis. Marrow hypercellularity is thought to predispose to this phenomenon by causing a decrease in local blood flow and oxygenation.
Dactylitis Dactylitis is a painful swelling of digits of the hands and feet (“hand-foot syndrome”). It occurs early in infancy as hematopoietic marrow is still present in these bones at this age. Most episodes resolve within in 2 weeks.227,228,229,230 Epiphyseal infarction can result in joint pain and swelling mimicking septic arthritis. Use of hydroxyurea in the BABY HUG trial resulted in significant reduction of rate of dactylitis.161
Osteomyelitis, Septic Arthritis, and Bone Infarction Impaired cellular and humoral immunity together with infarction of bone contribute to this complication with an estimated prevalence of 12 percent. Atypical serotypes of Salmonella, S. aureus, and Gram-negative bacilli are the principal infectious offenders. No single lab or imaging test reliably differentiates osteomyelitis from infarction.227,229,231,232,233,234,235 Culture results may be nondiagnostic as patients usually receive antibiotics on presentation with fever; therefore, the presence of leukocytes in bone and joint aspirates should evoke a high suspicion for osteomyelitis.126 Septic arthritis tends to occur in joints involved with avascular necrosis, also seen following hip arthroplasty. Multiple joints may be involved. An elevated C-reactive protein should raise suspicion for septic arthritis and prompt intervention with appropriate antibiotics as needed to prevent joint deterioration and collapse.227 Vertebral body infarctions with subsequent collapse causes the classic “fish mouth” appearance of vertebrae on radiographs of the spine.
Osteopenia and Osteoporosis Osteopenia and osteoporosis are prevalent (30 to 80 percent) in patients with sickle cell anemia, with a predilection for the lumbar spine. Presence of avascular necrosis with local bone remodeling may lead to false-negative results on a bone mineral density test at the femoral neck.126 Fractures of the long bones are commonly underdiagnosed and self-reported rates of fractures in young adults with SCD are high. Etiology of osteoporosis is multifactorial with hypogonadism, hypothyroidism, nutritional deficiencies, and iron overload interfering with osteoblast function being the major causes.126,236,237,238 More than 50 percent of patients are vitamin D deficient with the majority (>80 percent) having less-than-optimal levels. High doses of vitamin D supplementation have resulted in improvement in chronic pain and higher levels of physical activity.239
Avascular Necrosis Vasoocclusion resulting in infarction of articular surfaces of long bone occurs most commonly in the femur followed by the humerus. It was previously thought to occur with increased frequency in HbSC disease as opposed to patients with HbSS. However, with increased longevity of HbSS patients, its prevalence is greater in patients with HbSS.240,241,242 As per the CSSCD estimates, 50 percent of patients by age 33 years will have avascular necrosis of the femoral head (Fig. 49–9
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


