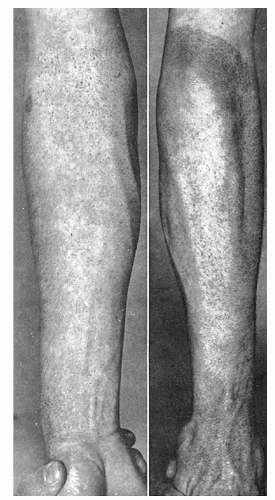
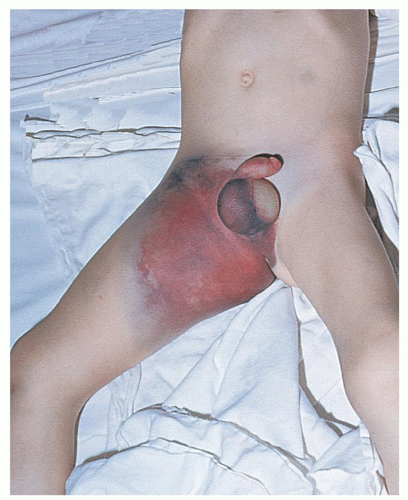
Bleeding Time
Hemostasis in a small superficial wound, such as that produced when measuring the bleeding time, depends on the rate at which a stable platelet plug is formed and, thus, provides a measure of the efficiency of the vascular and platelet phases. However, it does not discriminate between vascular defects, thrombocytopenia, and platelet dysfunction. The bleeding time leaves much to be desired in terms of reproducibility, because no two skin areas are exactly the same and it is impossible to produce a truly standard wound.
7Older studies using the bleeding time test supported the view that this test might be helpful in predicting bleeding in individual patients.
8 More recent studies suggest that a bleeding time result is determined not only by platelet number and function, but also by hematocrit,
9 certain components of the coagulation mechanism,
10,
11 skin quality,
12 and technique.
13 A careful analysis of this literature indicates that there is no correlation between a skin template bleeding time and certain visceral bleeding times,
13,
14 and that no correlation exists between preoperative bleeding time results and surgical blood loss or transfusion requirements.
15A clinical outcomes study reported that discontinuation of the bleeding time in a major academic medical center had no detectable adverse clinical impact.
16 A position paper of the College of American Pathologists and the American Society of Clinical Pathologists concluded that the bleeding time was not effective as a screening test, and that a normal bleeding time does not exclude a bleeding disorder.
17 Patients thought to have a platelet-type bleeding disorder based on their personal or family history (or both) should be evaluated for vWD and the inherited qualitative platelet disorders, using assays discussed in the section Platelet Function Assays. Newer assays that may be useful in screening patients for platelet dysfunction are also discussed in the section New Assays of Platelet Function.
Platelet Enumeration
Platelets are considerably more difficult to count than erythrocytes or leukocytes. This difficulty is to be expected in view of the small size of these cells and their tendency to adhere to foreign surfaces and to aggregate when activated.
In general, techniques for platelet counting may be classified into two groups: hemacytometer or direct methods, in which whole blood is diluted and the platelets are counted in much the same way as leukocytes or erythrocytes, and fully automated electronic methods. Virtually identical values for the normal range of the platelet count have been obtained with modern methods, as summarized in
Table 45.2.
An estimate of platelet numbers in a well-prepared blood smear by an experienced observer is a valuable check on the platelet count as determined by any method. In general, when a blood smear is examined at 100 × power, each platelet counted/field represents approximately 10,000 platelets × 109/L. Consequently, a normal blood smear should demonstrate, on average, at least 14 platelets/high-power field.
Instruments for totally automated platelet counting are widely used. Details of automated cell counters are discussed in
Chapter 1. When automated methods are used, various nontechnical factors may produce falsely low platelet counts.
18 These factors include platelet agglutinins,
19 abnormal amounts of plasma proteins in various paraproteinemias, previous contact of platelets with foreign surfaces such as dialysis membranes,
20 large or giant platelets, platelet satellitism,
21 lipemia,
22 and ethylenediaminetetra-acetic acid (EDTA)-induced platelet clumping,
23 a phenomenon that may produce platelet clumps of sufficient size to artifactually increase the leukocyte count.
24 Spuriously high platelet counts may result from the presence of microspherocytes,
25 fragments of leukemic or red blood cells,
26 and Pappenheimer bodies.
27 Special technical modifications and the use of careful manual counting methods may be required to eliminate these artifacts and to obtain accurate platelet counts.
Platelet Volume Measurements
The widespread availability of particle counters in the clinical laboratory permits the accurate measurement of platelet volume on a routine basis. Mean platelet volume (MPV) is increased in disorders associated with accelerated platelet turnover as the result of large numbers of megathrombocytes
28 or in patients with Bernard-Soulier syndrome. Normal or decreased values for MPV usually are obtained in patients with disorders associated with deficient platelet production, in some patients with sepsis,
29 and in people with certain big-spleen syndromes.
30Some authors suggest that increased MPV provides evidence of accelerated platelet production and may be interpreted in the same way as the reticulocyte count. The method is difficult to standardize, however, and when determined on routinely collected specimens by automated counters, it is affected by numerous variables pertaining to specimen collection, anticoagulant, temperature, and duration of storage.
31 In view of these problems and the difficulty in interpreting platelet size heterogeneity under normal and abnormal conditions,
32 these measurements should be interpreted with caution.
The presence of microcytic platelets in patients with some inherited thrombocytopenias such as Wiskott-Aldrich syndrome is reliably reflected by MPV measurements. On the other hand, giant platelets associated with Bernard-Soulier syndrome may be counted as leukocytes or erythrocytes and may not be reflected in the MPV.
Platelet Function Assays
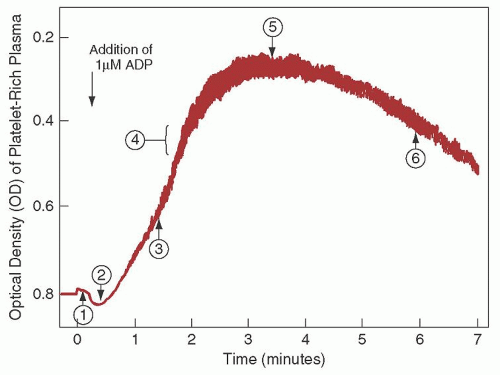
Since the 1960s, platelet aggregation using platelet-rich plasma has been the standard method to assess platelet function. This method uses aggregometers, which are modified nephelometers that permit measurement of changes in optical density of a platelet suspension under conditions of constant temperature and continuous agitation (
Fig. 45.3). Most instruments measure
a combination of light scatter and absorption. Instruments have been developed that permit both nephelometric and photometric measurements and the simultaneous measurement of aggregation and nucleotide release.
33Platelet aggregation usually is studied in suspensions of citrated platelet-rich plasma, in which the size and dimensions of the stirring bar, variations in plasma citrate concentration attributable to variations in hematocrit, the pH, and the nature of the buffers are important variables. Platelet suspensions usually are prepared by differential centrifugation, but methods that use albumin density gradient centrifugation and gel filtration have also been described.
34 Although harvesting platelets from the blood of thrombocytopenic patients is difficult, testing such platelets in the aggregometer is reproducible in suspensions containing as few as 50,000 platelets/
µl. Methods for the study of platelet aggregation in whole blood
35,
36 also have been described. Interfaced computer systems have been developed for calculating and expressing platelet function data.
37Adenosine diphosphate (ADP) in concentrations of 5
µmol/L or higher produces platelet aggregation directly that is independent of the release of platelet-contained ADP.
38 Various other aggregating agents act mainly by inducing the release reaction, such as a suspension of connective tissue particles (collagen), epinephrine and norepinephrine, and thrombin. With epinephrine (5
µmol/L), a weak primary aggregating effect usually can be clearly distinguished from the subsequent release reaction, which produces a secondary wave of aggregation. Such primary and secondary waves of aggregation also may be seen with carefully titrated amounts of ADP (0.2 to 1.5
µmol/L).
38Ristocetin is an antibiotic that induces platelet agglutination (platelet metabolic activity not required) in the presence of von Willebrand factor (vWF). Patients deficient in vWF (vWD) or in the receptor for vWF (Bernard-Soulier syndrome) have an abnormal ristocetin response. Ristocetin is tested in concentrations of 0.6 to 1.2 mg/ml; the lower concentrations are helpful in identifying specific variants of vWD, type 2B and platelet-type vWD (see
Chapters 52 and
53).
The release reaction is measured only indirectly by routine aggregometry—that is, the aggregation associated with the release of ADP from the platelets (release-induced aggregation or secondary aggregation). Methods for the quantitation of various substances released from platelets have been described. For example, the amounts of ADP or serotonin released/unit of time serve as indices of dense body release
39; the amount of various hydrolytic enzymes or platelet factor 4 released is a measure of the extent of
α-granule release.
40 Suggested guidelines for standardization of platelet aggregation methods have been proposed.
41,
42Sensitive methods have been developed for the determination of platelet-derived substances in plasma that may serve as markers of intravascular platelet activation,
43 including platelet factor 4,
β-thromboglobulin, stable prostaglandins (6-keto prostaglandin F
1α and thromboxane A
2), and leukotrienes.
43 These measurements may have diagnostic value in thromboembolic disorders and syndromes characterized by intravascular platelet aggregation.
New Assays of Platelet Function
An appreciation of the limitations of the bleeding time test has led to the development of newer assays to evaluate platelet function.
44 Some of these are point-of-care tests. The clinical use and predictive value of these tests to identify patients with hemostatic disorders remain to be established. One assay, the platelet function analyzer (PFA-100), has been investigated for several years, and many published reports using this assay are available. In this method, citrated blood samples are exposed to high shear rates in a capillary flowing through an aperture within a membrane coated with collagen and either ADP or epinephrine.
45 The closure time to hemostatic plug formation within the aperture is the endpoint of the test. A large study using the PFA-100 found that prolonged closure times could be attributed to specific quantitative or qualitative abnormalities in platelet function or vWF (or both) in 93% of patients tested.
46 However, the International Society on Thrombosis and Haemostasis has taken the position that the PFA-100 is insufficiently sensitive and specific to be used as a screening device for platelet disorders.
47 It has been suggested that optimal use of the PFA-100 in evaluation of hemostasis would use an algorithmic approach, evaluating not only PFA-100 closure times, but also a complete blood count, blood smear, and assays for vWD and platelet aggregation to further evaluate abnormal closure times. A recent addition to the PFA repertoire is the INNOVANCE PFA P2Y test designed to assess P2Y12-receptor blockade.
Several additional platelet function analyzers are available on the market, though they are not as well studied as the PFA-100.
48 The ICHOR II-Plateletworks system (Helena Laboratories, Beaumont, TX) compares impedance-derived platelet counts in samples with and without added platelet agonists to assess platelet function. This system has historically been used to evaluate cardiopulmonary bypass patients, but more recently has been applied to patients undergoing coronary stent placement. The Impact-R (Matis Medical, Beersel, Belgium) is an automated cone-and-plate research analyzer that assesses platelet adhesion and aggregation on a polystyrene surface under laminar flow conditions. The VerifyNow system analyzes platelet agonist-induced aggregation of fibrinogen-coated microparticles to assess platelet function. Agonist cartridges are designed to evaluate the effects of aspirin, clopidogrel, and GPIIb/IIIa platelet receptor inhibitor administration on platelet function. Platelet mapping, a modification of thromboelastography, measures the platelet contribution to clot strength in the presence of specific agonists.
49 To date, no platelet function analyzer assay has been sufficiently studied or validated to warrant routine clinical use.
48,
50,
51