Diagnostic Approach to Malignant and Nonmalignant Disorders of the Phagocytic and Immune Systems
Diagnostic Approach to Malignant and Nonmalignant Disorders of the Phagocytic and Immune Systems
Daniel A. Arber
Thomas L. McCurley
John P. Greer
Cost-effective, efficient evaluation of patients with hematologic diseases is often based on the quality of collaboration between clinicians and pathologists. This has become even more important as the number and complexity of diagnostic tests has exploded. Flow cytometry, cytogenetic, fluorescence in situ hybridization (FISH), and molecular genetic testing present opportunities for improving diagnostic precision but are expensive, and each has its limitations and shortcomings. The goal of this chapter is to guide selection of testing to minimize the cost and time involved in making diagnoses in common clinical problems in hematology.
This chapter introduces an approach to disorders that produce abnormalities of circulating blood cells, constitutional symptoms, or enlargement of lymph nodes or the spleen. The emphasis is on distinguishing benign from malignant disease. The disorders may take the form of (a) immune reactions, particularly infection resulting in changes in blood leukocytes, lymphadenopathy, and/or organomegaly; (b) neoplastic diseases (leukemias, lymphomas, and plasmacytic neoplasms and related diseases); and (c) inherited or acquired diseases that result in immunodeficiency.
APPROACH TO DIAGNOSIS
Patients with disorders of the immune system (innate and adaptive) usually come to medical attention with symptoms and signs suggestive of infection. Many patients with hematopoietic lymphoid neoplasms may also present with fever or other nonlocalizing constitutional signs and symptoms such as fatigue, generalized lymphadenopathy, abnormal bleeding, weight loss, bone pain, arthralgias, and pruritus. Suspicion of an underlying immune disorder, benign or malignant, may be raised during the course of a routine examination or during the evaluation of unrelated disorders by the detection of skin lesions, lymphadenopathy, splenomegaly, or hilar or mediastinal masses. In other instances, routine blood examination may disclose abnormalities in the numbers or morphology of circulating red cells, white cells, or platelets.
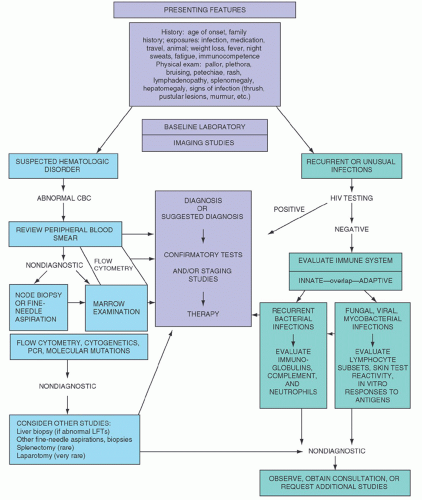
A detailed history should be obtained, and a thorough physical examination should be performed. Age of the patient, immunocompetence (focusing on types and number of infections), family history, travel history, animal exposure, history of prior therapy, and drug ingestion are particularly important areas to address. Complete blood cell count (CBC) and blood examination (including morphologic assessment of red cells, white cells, and platelets) may be diagnostic or help determine the next step in diagnosis. Chest x-ray and computed tomography (CT) may assist in identifying possible biopsy sites. Electrophoresis to characterize serum or urine immunoglobulins, marrow examination, lymph node aspiration or biopsy, liver biopsy, and splenectomy with flow cytometry immunophenotyping of blood, marrow, or tissue, may be necessary. In addition, evaluation of delayed type hypersensitivity and tests of neutrophil and/or lymphocyte function may be required. Finally, cytogenetic and molecular genetic testing may be necessary for a complete assessment. Clearly, not all or even most of these tests and surgical procedures are indicated for each patient who has the symptoms or signs described. Rather, a logical sequence is undertaken for each patient, tailored to the particular findings. A diagrammatic summary of a sequential approach is shown in Figure 56.1.
The examination of stained tissues by light microscopy remains the principal means of establishing a diagnosis in most of the benign and malignant conditions considered in Parts VI (Nonmalignant Disorders of Leukocytes, the Spleen, and/or Immunoglobins) and VII (Hematologic Malignancies). Other studies, however, including immunophenotypic studies, cytogenetics, and molecular genetics, are increasingly valuable in making, confirming, and “fine-tuning” diagnoses as well as in defining clinically important subcategories of disease.1, 2
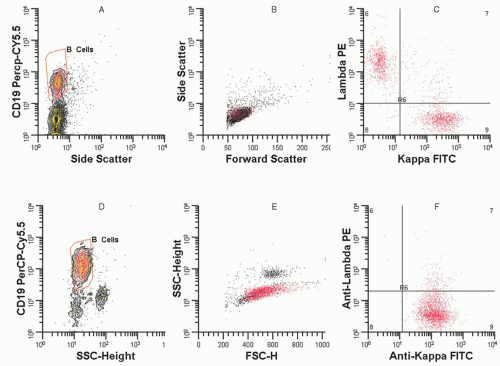
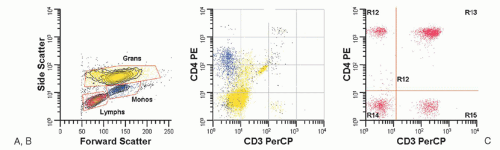
Flow cytometry has become a primary immunophenotyping method in the diagnosis, classification, and detection of residual disease in patients with hematologic malignancies, and is covered in more detail in Chapter 2. For example, flow cytometry is helpful in the differential diagnosis of reactive and neoplastic lymphoid proliferations, but requires specimens that are submitted fresh or in saline. Figure 56.2 demonstrates the difference between immunoglobulin light chain distribution in a patient with follicular hyperplasia (with polytypic light chain expression) versus a patient with a diffuse large B-cell lymphoma (with monotypic light chain expression). In T-cell neoplasms, the detection of a population of lymphocytes with aberrant phenotypes (e.g., lost or diminished expression of pan T-cell markers) as illustrated in Figure 56.3, is very helpful in detecting abnormal T-cell populations. Flow cytometry is essential for classification of acute leukemia, in part because most diagnostic studies can be completed in less than 2 hours. Additionally, flow cytometry immunophenotyping detects myeloid antigen expression in cases that are myeloperoxidase or Sudan black B negative by cytochemical studies. Finally, flow cytometry can detect aberrant blast cell immunophenotypes, which are useful for the detection of minimal residual disease in post-therapy specimens. The level of detection of minimal residual disease by flow cytometry is well below what can be identified by morphologic assessment of aspirate smears. However, flow cytometry does have limitations. It requires fresh or viable tissue and is of less value in the diagnosis of Hodgkin lymphoma (HL), T-cell-rich B-cell lymphomas, and subclassification of reactive processes (see Table 56.1), and may miss some T-cell neoplasms that do not demonstrate aberrant immunophenotypes. Most laboratories do not use methods that are effective in detecting the rare tumor cells of HL, but some studies have used this method effectively and this application of flow cytometry may emerge in the future.3 Additional applications in morphologically difficult areas such as myelodysplastic syndromes are evolving.
FIGURE 56.1. Diagnostic steps in diseases of the hematopoietic lymphoid system. CBC, complete blood cell count; HIV, human immunodeficiency virus.
FIGURE 56.2. Flow cytometry of reactive B cells and monotypic B cells. A: Histogram (picture) of pan B-cell anti-CD19 antibody expression versus this log side scatter signal (related to cellular complexity), which is used to define and identify the B-cell lymphocytes. B: Histogram shows lymphocytes based on forward scatter signal (size) versus log side scatter signal. The red population shows that the CD19+ B cells and predominantly T-cell CD19− population (black dots) are of similar small size. (1) C: A two-parameter histogram gating on CD19+ B cells with anti-Kappa fluorescein isothiocyanate (FITC) versus anti-lambda phycoerythrin. This pattern, which shows a mix of kappa and lambda, is consistent with a reactive polyclonal B-cell population. D: Histogram (picture) of anti-CD19 antibody expression versus log side scatter in a second patient. E: Histogram shows lymphocytes based on forward scatter signal versus log side scatter signal. The red population which represents the CD19+ B cells is larger in size than the smaller CD19− population (black dots). F: A two-parameter histogram of anti-kappa fluorescein isothiocyanate (FITC) versus anti-lambda phycoerythrin. Red dots are the CD19+ B cells from histogram and show a dominant kappa expressing B-cell population consistent with a large B-cell lymphoma. (Diagrams courtesy of Bruce Grieg, MT, Vanderbilt University.)
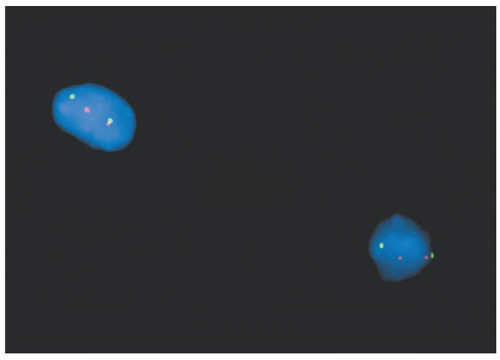
Cytogenetic studies have made enormous contributions to our understanding of the genetic pathogenesis of hematologic malignancies, and the field has established itself as a primary player in the diagnosis of prognostically important subsets of leukemia and lymphoma (e.g., Burkitt lymphoma, mantle cell lymphoma, and acute promyelocytic leukemia) (see Chapter 3). FISH has an expanding role in diagnosis (Table 56.2), and has several key advantages over routine cytogenetics: (1) it can be performed on interphase nuclei in fixed tissues (including paraffin-embedded), (2) results are available in 24 to 48 hours, and (3) it shows enhanced sensitivity over routine cytogenetics for critical translocations. Figure 56.4 illustrates FISH studies showing a MYC break apart probe in a patient with an 8;14 translocation in Burkitt lymphoma.
Molecular genetic studies, primarily polymerase chain reaction (PCR) techniques, are the most sensitive for detecting small critical genetic mutations or translocations in samples which may contain very few (less than one in a million) tumor cells (see Chapter 4). PCR has also replaced Southern blot analysis as the standard for the demonstration of T-cell and B-cell clonality and is ideal for detecting minimal residual disease in patients with lymphoma as well as in patients with chronic myelogenous leukemia (CML) (Table 56.3). Many of these applications work in fixed tissues. These studies are of limited or no value in patients with HL, myelodysplasia (MDS), or translocations that have widely spaced, variable breakpoints, as in mantle cell lymphoma.
Hematologic neoplasms are clonal tumors, and methods that can prove clonality of a cell population are extremely useful when the differential diagnosis is between a benign and neoplastic proliferation. The demonstration that an expanded B-cell population is clonal by immunoglobulin light chain restriction, karyotype analysis, or gene rearrangement studies facilitates the distinction between benign and malignant proliferations. However, clonality does not always indicate malignancy, and small clonal populations and pseudoclones may exist in non-neoplastic conditions.1, 2, 4, 5 Clinicians have recognized for years that the vast majority of patients with monoclonal gammopathies do not have an underlying B-cell neoplasm. Similarly, sensitive molecular genetic techniques frequently identify oligoclonal and monoclonal T- and B-cell populations in “benign” disorders, including lymphomatoid papulosis, autoimmune disorders (e.g., Sjögren syndrome and Hashimoto thyroiditis) in association with viral infections, celiac disease, and other lymphoid hyperplasias (particularly those in extranodal sites)5, 6, 7 (Table 56.4). Although these disorders may evolve into frank malignancy, they can remain clinically benign, remit spontaneously, or regress in the setting of antibiotics and removal of an antigen (Helicobacter pylori in gastric marginal zone lymphoma). Thus, although new techniques to identify clonality have astonishing sensitivity, the clinical, laboratory, and histopathologic data must be correlated to distinguish benign from malignant disorders. A major pitfall in diagnosis is failure to recognize that very sensitive studies for clonality by flow cytometry, protein electrophoresis, or molecular techniques are accompanied by a corresponding loss of specificity for the diagnosis of frank neoplasia.
FIGURE 56.3. Histograms demonstrating a phenotypically aberrant T-cell population in a patient with peripheral blood involvement by a T-cell lymphoproliferative disorder. In A, a side scatter versus forward histogram, the lymphocytes (low forward scatter, low side scatter) are painted red. The granulocytes with high side scatter and high forward scatter are painted yellow and the monocytes with forward scatter similar to granulocytes but with intermediate side scatter are painted blue. B displays the granulocytes and monocytes stained with anti-CD3 and anti-CD4, both of which do not stain with the pan T-cell marker anti-CD3 but the monocytes express intermediate levels of CD4. In C which displays CD3 and CD4 expression on the lymphocytes (painted red), normal T helper cells express CD4 and CD3 while the neoplastic T-cell population (upper left) expresses bright CD4 but have lost CD3. (Histograms courtesy Bruce Grieg, MT, Vanderbilt University.)
In the next sections, the examination of the blood, marrow, nodes, and spleen are reviewed in the context of making a diagnosis, introducing the subsequent chapters, and outlining overviews on the evaluation of fever of unknown origin (FUO) and patients with recurrent infection.
TABLE 56.1 MAJOR CLINICAL APPLICATIONS OF FLOW CYTOMETRY
Established
Evolving
Limited Value or Superceded by Other Techniques
Acute leukemia (diagnosis and detection of residual disease)
Major disadvantage of this technique: Requires viable cells. False negatives occur with necrotic or sclerotic tissues, partial tissue involvement and in tumors where neoplastic cells are in a minority (e.g., T-cell-rich B-cell lymphoma).
Major advantages of this technique: Can be applied to any tissue or body fluid. Rapid turnaround times.
aFlow cytometry should be performed on all fine-needle aspirations and tissue biopsies in which a hematopoietic lymphoid process is suggested by clinical history or touch preps/frozen sections.
bFlow cytometry using cytoplasmic immunoglobulin light chain markers is effective in establishing clonality of plasma cells in multiple myeloma, but not useful in accurately enumerating plasma cells.
TABLE 56.2 COMMON CLINICAL APPLICATIONS OF CYTOGENETICS INCLUDING FLUORESCENCE IN SITU HYBRIDIZATION
Acute Leukemia with prognostically important translocations
APL with t(15;17)(q24.1;21.1) (PML/RARA) and RARA variants
AML with t(8;21)(q22;q22) (RUNX1-RUNX1T1)
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22) (CBFB/MYH11)
AML with t(9;11)(p22;q23); MLLT3-MLL and other MLL translocations
ALL with t(12;21)(p13;q22) (ETV6-RUNX1)
ALL with t(4;11)(q21.3;q23) (AFF4-MLL) and other MLL translocations
ALL with t(9;22)(q34;q11.2) (BCR-ABL1)
Non-Hodgkin lymphoma with diagnostically critical translocations
Burkitt lymphoma: t(8;14)(q24;q32.33) (MYC-IGH@) and other MYC variants
FIGURE 56.4. FISH studies demonstrating aMYCtranslocation in Burkitt lymphoma using a dual colorMYCbreak apart probe with the red labeled probe binding the 5 prime end ofMYCand the green labeled probe binding the 3 prime end ofMYC. In the chromosomes with unarranged MYC, the green and red signals are together but the colors separate when MYC is part of a translocation with the IGH@ gene on chromosome 14.
EXAMINATION OF THE BLOOD
Physicians are too busy to examine peripheral blood smears in all of their patients. They usually do so in one of these circumstances: (a) patients with constitutional symptoms, particularly fever (see section on “Fever of Unknown Origin”) or weight loss, without an obvious underlying cause; (b) an abnormality in blood counts, either cytosis or cytopenia; or (c) as a part of the evaluation of a patient with clinical features of immunodeficiency.
The blood counts and peripheral smear are frequently abnormal in patients with constitutional symptoms, immunodeficiency, hematologic neoplasms, or a variety of other syndromes associated with lymphadenopathy or splenomegaly. Anemia, when present, usually is normochromic and normocytic. Many patients with chronic disease, MDS, plasma cell myeloma, and, rarely, acute leukemias have anemia but no other abnormalities. In patients with anemia of unknown origin, the evaluation begins with examination of the blood smear, red cell indices, and reticulocyte count. The evaluation proceeds based on the results of these simple tests as outlined in Chapter 22.
TABLE 56.3 COMMON APPLICATIONS OF NON-FLUORESCENCE IN SITU HYBRIDIZATION MOLECULAR GENETIC TECHNIQUES
Non-Hodgkin lymphoma with translocations with widely spaced breakpoints (e.g., mantle cell lymphoma)
T-cell clonality
TRG@ PCR
TRB@ PCR
Acute leukemia
Balanced translocation PCR (similar to those listed in Table 56.2 for FISH)
NPM1, FLT3, KIT, CEBPA, DNMT3A mutations
Myeloproliferative neoplasms
BCR/ABL1 PCR
JAK2 mutations
Post-transplant engraftment
aEmerging mutations may become prognostically significant in myelodysplastic syndromes.
TABLE 56.4 EXAMPLES OF CLONAL PROCESSES WHICH MAY BE CLINICALLY IRRELEVANT
B-Cell Clonality
Monoclonal gammopathies on protein electrophoresis with immunofixation
Small circulating monotypic B-cell populations in healthy adults by flow cytometry
Transient clonal IGH@ rearrangements by PCR in(1) salivary glands in Sjögren syndrome, (2) in thyroid tissues in Hashimoto thyroiditis, (3) in bone marrow following solid organ or bone marrow transplantation, (4) small biopsies (i.e., skin, gastric)
Clonal IGH@-BCL2 rearrangements detected in low quantity in normal children and adults in blood and tonsil by ultrasensitive PCR methods
T-Cell Clonality
Transient clonal T-cell receptor rearrangements by PCR in blood of healthy elderly patient
Clonal T-cell receptor rearrangement by PCR in skin of patients with reactive dermatitis
Clonal T-cell receptor rearrangement by PCR in celiac disease
PCR, polymerase chain reaction.
Abnormal Platelet Counts
Thrombocytopenia
The major differential diagnosis of patients with thrombocytopenia includes (a) thrombocytopenia secondary to autoantibody-mediated platelet destruction (idiopathic thrombocytopenic purpura [ITP], systemic lupus erythematosus [SLE], drug-related thrombocytopenia, and perhaps human immunodeficiency virus [HIV]); (b) primary marrow disease, particularly MDS or acute leukemia; and (c) peripheral consumption secondary to intravascular activation of clotting (e.g., disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, or hemolytic uremic syndrome). Although thrombocytopenia can be secondary to a nutritional deficiency (e.g., folate, vitamin B12), usually the macrocytic anemia is the dominant clinical feature.
In ITP and drug-related thrombocytopenias, the peripheral smear red cell and white cell morphology should be normal and rare giant platelets (megathrombocytes) are present. Thrombocytopenic patients with SLE may also be neutropenic and lymphopenic. Thrombocytopenia is present in up to 50% of patients with antiphospholipid antibody syndrome.8 Thrombocytopenia is the first sign of HIV infection in 10% of patients who may also be lymphopenic.9 Thrombocytopenia occurs in up to 45% of patients with hepatitis C viral infection. MDS and leukemia rarely present with isolated thrombocytopenia. Most MDS patients are anemic and have white blood cell (WBC) abnormalities (e.g., blasts or hyposegmented or hypogranulated polymorphonuclear neutrophils [PMNs]) that indicate primary marrow disease. Schistocytes and polychromatophilic red cells in patients with disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, and hemolytic uremic syndrome are helpful in pointing away from marrow disease or an autoimmune thrombocytopenia. In rare patients, platelets will satellite around PMNs or clump in blood collected in ethylenediaminetetraacetic acid. This artifact, readily apparent on examination of stained blood smears, disappears when blood is anticoagulated in heparin.10 Similarly, when blood is collected for platelet counts on heel sticks, clumping (also seen on smears) may produce spurious thrombocytopenia.
Congenital thrombocytopenias are rare, but can be misdiagnosed as ITP, and size of platelets may guide further evaluation.11 Small platelets in a male suggest X-linked Wiskott Aldrich syndrome (WAS). Large platelets are found in von Willebrand 2B, May-Hegglin anomaly, Bernard-Soulier syndrome, and the gray platelet syndrome. Normal-sized platelets are found in amegakaryocytic thrombocytopenia, thrombocytopenia with absent radii, and familial platelet disorders with a predisposition to acute myeloid leukemia (AML).
Drug-induced thrombocytopenia (DITP) may be misdiagnosed as ITP and overlooked if patients are not asked about over-thecounter medicines, food, beverages, and/or herbal supplements.11 DITP can be due to immune-mediated or non-immune processes. In a large series of patients initially diagnosed as ITP, 8% were found to have DITP with quinine (including tonic water) as the most common cause.12 Hematologists are frequently consulted about heparin-induced thrombocytopenia (HIT) which is due to antibodies against platelet factor 4 heparin complexes and leads to an increased risk of thrombosis.13
Bone marrow examination with cytogenetic studies is essential only in the cases of thrombocytopenia in patients with suspected MDS or acute leukemia or in those for whom the diagnosis of autoimmune or consumptive thrombocytopenia is in doubt. In most children or young adults with otherwise normal counts and a normal blood smear, a bone marrow is probably not needed.14 Amegakaryocytic thrombocytopenias secondary to genetic or autoimmune disease are exceedingly rare.
Thrombocytosis
Thrombocytosis with platelet counts of greater than 600 × 109/L are commonly seen transiently during a variety of infections, after splenectomy, after acute blood loss, or during recovery from marrow injury or nutritional deficiency. The concern in persistent thrombocytosis is the presence of an underlying myeloproliferative neoplasm or occult malignancy. The presence of a myeloproliferative neoplasm is almost always suggested by the peripheral blood counts and smear (e.g., polycythemia in polycythemia vera [PV], leukoerythroblastic changes in primary myelofibrosis [PMF or agnogenic myeloid metaplasia], and leukocytosis with immature myeloid precursors in CML). The one myeloproliferative neoplasm in which thrombocytosis is often an isolated finding is essential thrombocythemia (ET), a diagnosis that is supported by the demonstration of megakaryocytic dysplasia on bone marrow examination or the demonstration of JAK2 mutations on molecular genetic studies of peripheral blood.15, 16 The JAK2 V617F mutation is estimated to occur in 60% of ET and myelofibrosis and 95% of PV.17 Congenital thrombocytosis is rare and results from mutations in either thrombopoietin (THPO) or its receptor (MPL).18
Morphologic Changes in Leukocytes
Morphologic changes in WBC are often diagnostic of certain rare diseases such as Alder-Reilly or Pelger-Huët anomaly, some of which are associated with infection (Chediak-Higashi syndrome) (Chapter 58).
Changes in the blood of patients with acute leukemia strongly suggest the diagnosis in virtually all patients.7 In approximately 60% of these individuals, the smear confirms the diagnosis by the presence of a large proportion of immature cells, whether or not the leukocyte count is increased. Blasts are easily demonstrable in blood smears of more than 90% of patients, and, even in patients whose blood smears have few blasts, anemia, thrombocytopenia, neutropenia, or combinations of these changes are suggestive of acute leukemia. For almost all such patients, bone marrow aspiration proves diagnostic and should be supplemented by flow cytometry immunophenotyping and karyotype analysis.
Lymphocytosis of Small Mature Lymphocytes
More than 5.0 × 109 small lymphocytes/L of blood in adults over 50 years of age is usually indicative of chronic lymphocytic leukemia (CLL). Many patients are asymptomatic and one-fourth are diagnosed while undergoing blood work for another problem. Flow cytometry demonstrating small B lymphocytes with weak CD20 and weak, monotypic light chain expression and coexpression of CD5 and CD23 is characteristic. Proceeding with other examinations in such patients, such as lymph node biopsy or bone marrow examination, is usually unnecessary, but immunophenotyping is required to exclude other B-cell lymphomas in the blood, such as mantle cell lymphoma, that may mimic CLL. In children, pertussis is associated with lymphocytosis of small CD4 T cells19 that may be worrisome for a lymphoid leukemia on review of the blood smear. Chronic polyclonal B-cell lymphocytosis is well described in young to middle-aged asymptomatic women who usually smoke.20 These women typically present with a lymphocyte count that is less than 5 × 109/L, the number required for a diagnosis of CLL, and the B cells are polytypic by immunoglobulin light chain studies and by PCR studies of the immunoglobulin heavy chain. Monotypic B-cell proliferations that present with less than 5 × 109/L B cells are now classified as monoclonal B lymphocytosis rather than as overt CLL.21
Lymphocytosis of “Atypical Lymphocytes”
Atypical or abnormal lymphocytes are a classic feature of Epstein-Barr virus (EBV) infectious mononucleosis but also occur in other acute viral infections including cytomegalovirus (CMV), hantavirus, hepatitis, HIV, and less commonly in lymphomas with a leukemic phase (e.g., human T-lymphotropic virus -1, hepatosplenic T-cell lymphoma, etc.). Thrombocytopenia, an elevated hematocrit, neutrophilia, and circulating lymphoblasts in a patient from the desert Southwest strongly suggest hantavirus infection.22 Usually the clinical features and other routine laboratory tests such as serologic studies for viral infection make the diagnosis straightforward. When distinguishing reactive lymphocytes from leukemic blasts or the leukemic phase of a lymphoma is difficult, flow cytometry can provide invaluable help in defining the phenotype and clonality of circulating cells.
Neutrophilic Leukocytosis
The presence of CML is strongly considered in a patient with neutrophilic leukocytosis, particularly when the WBC count is greater than 50.0 × 109/L and is accompanied by an increase in circulating immature myeloid cells and basophilia. Additional findings suggesting the diagnosis of CML include (a) symptoms of fatigue, bone pain, left upper quadrant pain, or fullness; (b) sternal tenderness and splenomegaly on physical examination; and (c) thrombocytosis. A leukemoid reaction must also be considered and can be caused by either benign, particularly infectious, or malignant conditions. Interphase FISH studies on peripheral blood for the BCR/ABL1 translocation can establish the diagnosis of CML, obviating the need for bone marrow examination; but many recommend an initial bone marrow to screen for other cytogenetic abnormalities that could affect prognosis.
Chronic idiopathic neutrophilia is defined by leukocytosis of 11.0 to 40.0 × 109/L and is associated with smoking and obesity.23 Familial neutrophilia (WBC > 20.0 × 109/L) is a rare autosomal dominant disorder which is caused by a mutation in the granulocyte colony-stimulating factor (G-CSF) receptor.24 A transient myeloproliferative disorder is seen in up to 10% of children with Down syndrome.25 Neonates with persistent leukocytosis, delayed separation of the umbilical stump, and recurrent infections should be screened for leukocyte adhesion deficiency (LAD) (see section “Recurrent Infections”).
Monocytosis
Monocytosis (> 1.0 × 109/L) is commonly caused by infections, including granulomatous disease (tuberculosis [TB], fungal disease), brucellosis, infectious mononucleosis, syphilis, protozoal infections (kala-azar, malaria) and rickettsial infections. Monocytosis may be prominent in chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia, or AMLs with a monocytic component.26 These neoplastic monocytic proliferations cannot be distinguished on peripheral blood evaluation alone and a bone marrow examination is necessary to separate CMML from AML. Autoimmune disease, inflammatory bowel disease, and sarcoidosis may be associated with monocytosis. Monocytosis has been associated with carcinomas (lung, colorectal, renal) and has been caused by secretion of interleukin (IL)-6 by the neoplastic cells.27
Eosinophilia
Unexpected eosinophilia is most commonly seen in patients with atopy or reactions to drugs or environmental allergens.28 Autochthonous parasitic disease as a cause of eosinophilia is rare in the United States, but visceral larva migrans caused by Toxocara canis or cati can be seen. In international travelers and immigrants, evaluations for tissue invasive parasites is warranted. In patients who are immunosuppressed (particularly organ transplant recipients and patients on high-dose steroids for autoimmune diseases) strongyloides should be excluded.
Persistently high eosinophilia (> 1.5 × 109/L for > 6 months) carries significant risk for end organ damage and requires thorough investigation. A minority of these patients have elevated IL-5 levels with circulating clonal T-cell populations with aberrant phenotypes by flow cytometry.29 Several neoplastic eosinophil proliferations are now recognized (see Chapter 84), including a group with a cryptic translocation involving FIP1LI-PDGFRA.30, 31 The latter patients can respond to imatinib therapy, while other genetic subtypes do not.
Increased Circulating Plasma Cells
Patients with plasma cell (multiple) myeloma usually have a few plasma cells in the blood, although occasionally large numbers are noted (plasma cell leukemia). Such patients almost invariably have other symptoms and signs of myeloma. A few plasma cells may also be observed in blood smears of patients with infectious mononucleosis or other viral infections. Plasma cells also are seen in the blood of persons recovering from bacterial infections or having allergic reactions, especially serum sickness. The first step when the diagnosis of myeloma is considered is to perform studies of serum and urine immunoglobulins. The technique of immunoelectrophoresis has been replaced by electrophoresis with immunofixation. This sensitive and specific technique identifies a monoclonal serum Ig and/or light chain in urine in more than 5% of patients with myeloma. The diagnosis is confirmed on bone marrow examination.
Neutropenia
Neutropenia may be mild (ANC < 1.5 but > 1.0 × 109/L), moderate (ANC < 1.0 but > 0.5 × 109/L), or severe (ANC < 0.5 × 109/L).32 There can be ethnic variation, as exemplified by a prevalence of neutropenia of 4.5% among African-Americans in the United States compared to 0.79% and 0.38% in whites and Mexican-Americans, respectively.33 While infections and drugs are the leading causes of neutropenia, nutritional deficiencies (B12, folate, copper) should be ruled out early because of their responsiveness to replacement therapy. Alcohol abuse may present with isolated neutropenia, but is usually accompanied by other features of alcohol or nutritional marrow injury (macrocytosis, ringed sideroblasts). Immune-mediated neutropenia can be primary without an etiology or secondary to drugs, particularly B-lactam antibiotics and antithyroid medications; and autoimmune disease, including SLE, rheumatoid arthritis, and Graves’ disease.34, 35, 36 Late-onset neutropenia following rituximab therapy has been reported in 5% to 27% of lymphoma patients.37
Chronic idiopathic neutropenia (CIN) is a diagnosis of exclusion, rarely has increased infections, and is due to increased inflammatory changes within the bone marrow microenvironment. CIN is associated with activated T cells and increased levels of interferon γ (IFN-γ), tumor necrosis factor α, Fas-ligand, and transforming growth factor β1, which suppresses IL-10, an antiinflammatory cytokine, and promotes apoptosis of neutrophil progenitor cells.38
Hematologists should be familiar with congenital neutropenias, particularly cyclic neutropenia and severe congenital neutropenia (Kostmann syndrome), because the former can be mild and not be recognized until adulthood and the latter, which is supported by G-CSF, can evolve into leukemia in one-fifth of patients.39 Mutations in the neutrophil elastase gene (ELA2) account for 60% of cases of severe congenital neutropenia, either in autosomal dominant or sporadic forms, while mutations in the HS-1-associated protein X (HAX1) gene are found in 30% of cases and are autosomal recessive.40
Bone marrow aspiration and biopsy are usually reserved for neutropenia patients where there is an index of suspicion of malignancy usually due to immature cells in the differential; physical clues, such as adenopathy or splenomegaly; and/or other hematologic and laboratory abnormalities. MDS rarely presents as an isolated neutropenia and usually will have accompanying dysplastic abnormalities in more than one cell line. Patients with two indolent leukemias, hairy cell leukemia and large granular lymphocyte (LGL) leukemia, often present with bacterial infections secondary to neutropenia. Circulating hairy cell cases may be few in number, but a bone marrow examination with flow cytometry is usually diagnostic. LGL leukemia can be either T-cell (85% to 90%) or NK-cell (10% to 15%) in origin, is suspected when there are LGLs in the peripheral blood smear, and can be diagnosed by flow cytometry.41 Molecular genetic studies of the T-cell receptor gene can be confirmatory of clonality in T-LGL.42
Lymphopenia
Lymphopenia is defined as an absolute lymphocyte count < 1.5 × 109/L in adults and < 2.0 × 109/L in children. Since 70% to 80% of circulating lymphocytes are T cells (most of which are CD4 T cells), severe lymphopenia implies T-cell deficiency and warrants serologic investigation for HIV infection. In infants with infection, the possibility of severe combined immunodeficiency (SCID) or DiGeorge syndrome is suggested. Lymphopenia is common in systemic autoimmune diseases like lupus and sarcoidosis and in serious illness/infections associated with hypercortisolism.
A variety of viral infections including influenza and measles may produce transient lymphopenia in association with loss of skin test reactivity and transient susceptibility to reactivation of TB.43 One rare but noteworthy population with persistent lymphopenia is HIV-negative patients with CD4 T-cell counts less than 0.3 × 109/L. These patients fall into two categories, those susceptible to infection (who usually have low CD4/CD8 ratios) and those who have no increased susceptibility for infection with normal CD4/CD8 ratios.44
Peripheral Blood Findings in Infection
Blood leukocyte changes that are secondary to a variety of diseases usually consist of neutrophilia, eosinophilia, monocytosis, or abnormal-appearing lymphocytes. Changes in the distribution or appearance of circulating WBCs may provide important clues to the presence and type of an underlying infection. Eosinophilia is common in atopic individuals but can be seen in many parasitic infections. Atypical lymphocytosis is a helpful diagnostic clue in viral infections, particularly EBV infection. In many parasitic diseases, such as malaria, acute Chagas disease, and filariasis, the infectious agent may be demonstrated on peripheral blood smears. However, these three parasitic diseases are rarely autochthonous in North America, and the diagnosis is suggested by the nationality as well as the recent travel history of the patient.45, 46, 47
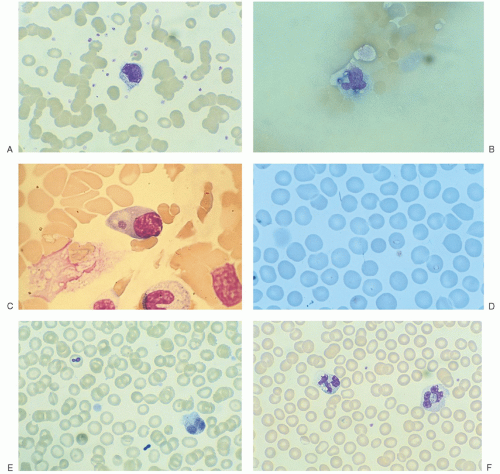
FIGURE 56.5. Wright-stained peripheral blood smears demonstrating (A)Mycobacteria avium-intracellulare as negative staining rods in a monocyte from patient with human immunodeficiency virus infection, (B)Histoplasma capsulatum with three intracellular yeast forms in a monocyte at the feather edge of smear in patient with acquired immunodeficiency syndrome, (C)Ehrlichia chaffeensis as a single basophilic cytoplasmic inclusion (photomicrograph courtesy of Dr. Charles Coleman, University of Missouri), (D) babesia as single intracellular ring forms in two erythrocytes, (E)Malassezia furfur as extracellular budding yeasts (one of which is overlying an erythrocyte) in an infant on total parenteral alimentation, and (F) intracellular bacteria in a neutrophil with an adjacent vacuolated neutrophil in a patient with septicemia.
Although in most infections the causative agents are not demonstrable in peripheral blood, there are certain notable exceptions. In acquired immunodeficiency syndrome (AIDS) patients or in other severely immunocompromised patients, mycobacteria (Fig. 56.5A), fungi, such as histoplasmosis (Fig. 56.5B), or Candida may occasionally be identified on the peripheral blood smear. Residents of the central United States (particularly Arkansas and Missouri) and the Southeast may acquire ehrlichiosis through tick vectors. Anaplasmosis, formerly called human granulocytic ehrlichiosis, is a closely related tickborne disease found more often in the Northeast and upper Midwest. The typical inclusions in mononuclear cells (Ehrlichia chaffeensis) (Fig. 56.5C) or in neutrophils (Anaplasma phagocytophilum) are present in up to one-third of these patients, who often present with unexplained fever with accompanying leukopenia and elevated transaminases. The diagnosis of ehrlichiosis/anaplasmosis may be confirmed either serologically or by PCR methods.48 Babesiosis, first recognized in the 1890s as the cause of Texas cattle fever, has now been identified in febrile patients in the Northeast (Nantucket, Martha’s Vineyard, Connecticut), Wisconsin, and on the West Coast. Infections can be life threatening in splenectomized patients. The diagnosis is suggested by finding single ring forms (1.0 to 3.5 µm in diameter) in red blood cells (Fig. 56.5D) (characteristic tetrads are usually seen only in infected animals).49 In nonimmunocompromised patients with indwelling venous catheters, Malassezia furfur (Fig. 56.5E), Candida, and Torulopsis (C. glabrata) may be identified in the peripheral blood.50, 51 In bacterial sepsis, visible circulating bacteria are rare, but the presence of cytoplasmic neutrophilic vacuoles in a fresh finger stick blood smear is a specific clue to the diagnosis of septicemia52 (Fig. 56.5F). Toxic granulation (also seen in patients with sepsis) is often quite prominent in uninfected patients receiving granulocyte or granulocyte/macrophage colony-stimulating factor.
EXAMINATION OF THE BONE MARROW
If a diagnosis is not established by examining the blood, bone marrow examination may be helpful (Table 56.5). Needle biopsy is usually performed in addition to aspiration, particularly if lymphoma, carcinoma, granulomatous conditions, or myelofibrosis are under consideration. To adequately triage marrow, the stained aspirate smear is reviewed along with the clinical information and any previous studies shortly after the procedure. This allows rapid decisions about cytogenetics (including FISH), molecular genetics, and flow cytometry to be made when viable cells are still available and also limits inappropriate studies (Table 56.6). In the case of inaspirable marrows, touch preparations should be made for morphologic assessment, and sufficient cells for flow cytometry may be obtained by maintaining negative pressure on withdrawal of the biopsy or aspirate needle and then rinsing the needle in nutrient media or buffered saline. In some cases, a second, fresh core biopsy can be minced and processed for flow cytometry and cytogenetic studies.
In cases of acute leukemia, the first question is whether it is lymphoid or nonlymphoid. Identification of Auer rods on Wright-stained aspirate smears is specific for a myeloid leukemia; however, Auer rods are found only in a minority of cases. Morphology is very important in the early identification of myeloid leukemias with promyelocytic differentiation, which can be sent for confirmatory FISH studies for the 15;17 translocation and can be completed in less than 24 hours. Flow cytometry is now a standard part of the diagnosis of many hematologic diseases, including all acute leukemias (Table 56.1), and for detecting minimal residual disease post therapy.53, 54, 55, 56, 57 Most cases (> 95%) can be classified as to lineage in less than 2 hours during the normal working hours of the laboratory. Flow cytometry is helpful in suggesting possible promyelocytic differentiation (DR−, CD2+, bright myeloperoxidase), megakaryoblastic differentiation (CD61+, CD41+, myeloperoxidase−), or the t(8;21)(q22;q22) (CD19+, CD34+, myeloid antigen+), but is not as useful in identifying other subsets in the French-American-British Cooperative Group (FAB) or World Health Organization classification. Cytochemical stains which were essential for FAB classification are now being performed infrequently and are now used most commonly when flow cytometry findings are ambiguous, especially in the case of possible mixed phenotype acute leukemia. Immunohistochemistry on the bone marrow core biopsy is helpful for lesions that may not be present in aspirated material, such as fibrosed areas of lymphoma or metastatic tumors. In some cases, both flow cytometry immunophenotyping and immunohistochemistry are required due to sampling differences between the aspirate and biopsy. Some cytoplasmic markers, such as BCL2 and cyclin D1, are only available by immunohistochemistry in most laboratories.
TABLE 56.5 INDICATIONS FOR BONE MARROW EXAMINATION
Unexplained cytopenia
Evaluation of leukemia
Confirmation of myeloproliferative neoplasm
Unexplained lymphadenopathy
Splenomegaly without a diagnosis
Diagnosis or staging of lymphoid neoplasms
Constitutional symptoms (fever, weight loss) without a diagnosis
Bone pain with abnormal laboratory work (e.g., complete blood cell count, ↑ protein, ↑ lactate dehydrogenase, and ↑ uric acid)
cIf flow cytometry shows an aberrant T-cell or monotypic B-cell population
dFlow cytometry underestimates the percentage of plasma cells in bone marrow which is better assessed by immunohistochemistry on tissue sections
FISH and cytogenetics are playing an expanded role in the classification of hematologic neoplasia and the identification of prognostically favorable and unfavorable subsets of disease.54, 58, 59, 60, 61 Favorable cytogenetics in AML include t(15;17)(q24.1;21.1) (PML/RARA) of APL, t(8;21)(q22;q22) (RUNX1-RUNX1T1), and inv(16) (p13.1q22) or t(16;16)(p13.1;q22) (CBFB/MYH11);these identify patients who are predominantly treated with either all-transretinoic acid plus chemotherapy (APL) or chemotherapy alone without early transplantation. Hyperdiploidy (more than 50 chromosomes per leukemia cell) and t(12;21)(p13;q22) (ETV6-RUNX1) are found in approximately 50% of childhood acute lymphoblastic leukemia (ALL) and confer a favorable prognosis.62 Hypodiploidy (less than 45 chromosomes per leukemia cell), t(4;11)(q21.3;q23) (AFF4-MLL), and t(9;22)(q34;q11.2) (BCR-ABL1) are associated with a poor prognosis in ALL.
In suspected MDS, FISH and cytogenetic studies are helpful when morphologic abnormalities in hematopoietic cells are relatively subtle.63, 64 Identifying 5q31 deletion in MDS is important because the patients may respond to lenalidomide.65 Patients with abnormalities of chromosome 7 and/or complex cytogenetics are more likely to evolve to AML and should be considered for early allogeneic transplantation. Flow cytometric evaluation of MDS is an evolving application which holds great promise, although it is not well standardized in this application and thus not available at many institutions.66 Flow cytometry is of no help in the evaluation of myeloproliferative neoplasms except when the presence of increased blasts on marrow smear point to an evolving acute leukemia/blast transformation.53
Molecular genetics can identify clonality in lymphoid neoplasms, confirm diagnoses in CML and subtypes of acute leukemia, and monitor minimal residual disease when a molecular marker is present (Table 56.3). NPM1 and CEBPA mutations, when present alone, are favorable mutations in AML while FLT3 and DNMT3 mutations are usually unfavorable. In the core binding factor leukemias (AML with t[8;21][q22;q22][RUNX1-RUNX1T1] and AML with inv[16][p13.1q22] or t[16;16][p13.1;q22][CBFB/MYH11]), mutations of KIT are usually unfavorable. JAK2 mutations are present in most patients with PV and over one-half of patients with PMF and ET.15, 17 Genomic profiles are being evaluated in hematopoietic neoplasms and can discriminate prognostic subgroups. Restriction fragment length polymorphisms and chimerism studies identifying percentage of donor myeloid and lymphoid cells are employed to monitor engraftments in allogeneic transplantation.
Lymphocytosis on a marrow smear is common in very young children. Phenotypically, the lymphocytes are usually B cells in all stages of differentiation (so-called hematogones). In such cases, the numbers and morphology of the myeloid, megakaryocytic, and erythroid cell lines are usually normal, which is helpful in differentiating these non-neoplastic precursor B cells from the blasts of the pre-B ALL.67 Lymphocytosis on marrow smear in an older adult should always have phenotypic studies by flow cytometry. Further cytogenetic/FISH studies are helpful in small B-cell lymphomas with ambiguous phenotypes or in confirmation of the diagnosis of mantle cell lymphoma (by FISH).68 In unusual proliferations, cells from the bone marrow aspiration should be frozen for possible future studies.
Bone marrow examination should also be considered in patients who complain of bone pain whether or not bone lesions have been demonstrated radiographically and particularly if there is an abnormal CBC or abnormal chemistries, such as an elevated lactate dehydrogenase (LDH), total protein, or uric acid. Some processes, including myeloma, lymphoma, carcinoma, neuroblastoma, and granulomatous infections, may be quite focal and missed on marrow examination. Occasionally, radionuclide scans with gallium-67 or indium,111 CT, magnetic resonance imaging (MRI), and/or positron emission tomography (PET) scans may be helpful in localizing various types of tumors and identifying specific marrow sites for biopsy.69, 70, 71, 72, 73 Again, serum and urine electrophoresis, with immunofixation (which allows detection of monoclonal immunoglobulins hiding in the beta region) should be performed on adults with bone pain. Screening tests for neuroblastoma, including urine catecholamines, should be performed in children with bone pain and a nondiagnostic marrow examination.
Only gold members can continue reading. Log In or Register to continue
Oct 21, 2016 | Posted by drzezo in HEMATOLOGY | Comments Off on Diagnostic Approach to Malignant and Nonmalignant Disorders of the Phagocytic and Immune Systems