Diabetes Mellitus and Wound Healing
Parham A. Ganchi
Elof Eriksson
Diabetes mellitus, the most common endocrine disease, is a complex systemic disorder with a broad range of effects on nearly every organ system. It ranks in the top ten causes of death in the United States and encompasses a heterogeneous and progressive group of abnormalities with a wide spectrum of severity (1). Although difficult to determine precisely because of inconsistent standards of diagnosis, the prevalence of diabetes in the United States is probably between 1% and 2%, with a range as high as 11% (2). Generally, it is clear that both morbidity and mortality are increased in this population. However, the effects of diabetes on wound healing remain controversial and the mechanism underlying altered wound healing is poorly understood.
Diabetes itself is one of the earliest described diseases. The Ebers Papyrus dating back to 1500 B.C. lists some of the symptoms and possible treatments for this ailment (3). The gangrenous foot wound is clearly described in biblical writings, although its association with diabetes was likely unknown. In 1887, Pryce describes the chronic painless foot ulcer associated with diabetic neuropathy (4). The diabetic foot ulcer continues to be a significant healthcare problem and a major source of morbidity for the diabetic patient. In fact, ulcers of the leg and foot are the most common complications of diabetes (5). Furthermore, there are estimates that diabetic ulcers will increase by 14% per year (6). Fifteen percent of all patients with diabetes will develop a nonhealing foot wound during their lifetime despite meticulous glucose and dietary control (7,8). Diabetic patients have a 15-fold higher risk of lower-extremity amputation and account for at least 50% of the more than 50,000 nontraumatic amputations performed in the United States each year (9,10). The financial cost of the diabetic foot wound to society is formidable. An estimated $500 million is spent annually, with amputations costing about $25,000 in the United States (11). In one study, successful conservative management of a diabetic foot ulcer led to a savings of 80% compared with the cost of amputation (12). Furthermore, these estimates do not include financial and psychological losses incurred by the diabetic patient, his or her family, and society as a result of the limitations imposed by the amputation.
The cliché that postoperative wound complications are increased in patients with diabetes is not entirely supported by the literature (13). If patients are matched for age, sex, weight, and comorbidities, several studies show that diabetic patients have no significant increase in postoperative wound complications as compared with nondiabetic patients (14,15). In fact, no increase has been found in postoperative cardiopulmonary, vascular, or infectious wound complications after major vascular or abdominal surgery (16,17,18,19,20). This is in sharp contrast to a number of animal and in vitro studies that clearly document that optimal glycemic control leads to optimal wound healing in diabetes (21,22,23,24,25). Hyperglycemia and insulin deficiency are linked to decreased formation of granulation tissue and collagen, poor deep-wound tensile strength, deficient capillary ingrowth, and defective immune function, among other deficiencies (22,26,27,28,29,30,31,32). Whether these laboratory findings have clinical significance remains to be shown.
The wound classically associated with diabetes is the nonhealing foot ulcer (Fig. 68.1). Wounds in other locations are rarely as challenging clinically, and few data exist to support an association between diabetes and wounds other than the foot ulcer. Elimination of the diabetic foot ulcer would reduce the number of in-hospital days for the diabetic patient more than the elimination of any other problem associated with diabetes. It is one of the most common and morbid complications of diabetes, and a great deal of literature has been written on this subject. To review the effects of diabetes on clinically relevant derangements in wound healing, we will examine the pathophysiology of the foot ulcer. Specifically, we will review the triad of neuropathy, ischemia, and infection, all common complications of diabetes and all culprits in the pathogenesis of the nonhealing foot ulcer.
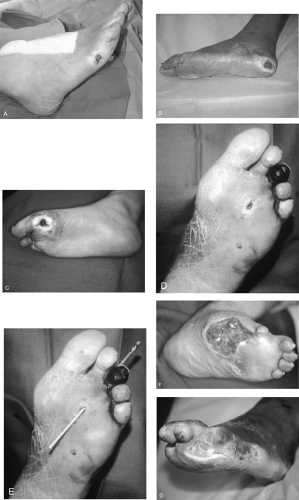 Figure 68.1. Typical diabetic foot ulcers. A: Early neuropathic ulcer over fifth metatarsal prominence. B: Chronic heel ulcer. C: Neuropathic forefoot ulcer over first metatarsal head with callus formation. D, E: Deceptively small metatarsal head ulcer with necrotic toe. Note true extent of ulcer, penetrating the foot to the dorsum. F, G: Advanced diabetic ulcers with extensive soft tissue and bone loss. Note clawing of toes. (See also color plate 3, color plate 4.) |
NEUROPATHY
Neuropathy in the diabetic patient can take many forms. They range from the “classic” chronic progressive distal symmetric neuropathies to the acute mononeuropathies to the pressure palsies most often affecting the median and ulnar nerves (33,34). The distal symmetric neuropathy is the most common symptomatic neuropathy associated with diabetes mellitus (35). It is a diffuse neuropathy with sensory loss predominating over motor deficits. The symptoms start distally and progress proximally in a stocking distribution, with the longest nerves affected first (36,37). Pathologically, there is a distal dying back of the axons (33,36). Mainly large myelinated fibers are lost,
with variable loss of small unmyelinated fibers (38,39,40). There is focal demyelination and regeneration with slowing of conduction velocities, elevation in sensory thresholds, and slowing of axonal transport (33,41,42,43). Signs and symptoms vary with the nerve fibers involved (44). Loss of large sensory fibers can lead to diminished light touch and proprioception, and damage to large motor fibers, though less common, may result in unsteadiness and weakness of the intrinsic muscles of the foot. Injury to small fibers results in increased pain thresholds and an inability to recognize temperature changes. These abnormalities predispose to repeated foot injuries and eventually to ulceration.
with variable loss of small unmyelinated fibers (38,39,40). There is focal demyelination and regeneration with slowing of conduction velocities, elevation in sensory thresholds, and slowing of axonal transport (33,41,42,43). Signs and symptoms vary with the nerve fibers involved (44). Loss of large sensory fibers can lead to diminished light touch and proprioception, and damage to large motor fibers, though less common, may result in unsteadiness and weakness of the intrinsic muscles of the foot. Injury to small fibers results in increased pain thresholds and an inability to recognize temperature changes. These abnormalities predispose to repeated foot injuries and eventually to ulceration.
The prevalence of neuropathy in the diabetic population has been difficult to determine, with estimates ranging from 10% to 90% (45,46,47,48,49). The Diabetes Control and Complications Trial (DCCT) quoted a 50% prevalence of significant neuropathy among their population, although a range of 20% to 30% represents more closely those patients who have symptoms or findings on examination (45,50). Generally, the incidence of neuropathy seems to increase with duration of disease and severity of hyperglycemia (51,52,53). One large study found an incidence of neuropathy of 8% at diagnosis of diabetes that increased to 50% at 25 years after diagnosis (54). However, this association is far from perfect. Many studies have shown a correlation between neuropathy and height, age independent of duration of diabetes, and smoking (45,51,54,55). Furthermore, the DCCT failed to find a correlation between neuropathy and levels of glycosylated hemoglobin (45).
Autonomic neuropathy also plays a role in diabetic foot ulceration and closely parallels the incidence of somatic neuropathy. Like somatic neuropathy, the incidence of autonomic neuropathy seems to increase with duration of disease. One study found a 4% incidence at the time of diagnosis of diabetes. This increased to 28% after 5 years (56). Others have shown a prevalence of 50% in unselected diabetic populations (57). Abnormal autonomic function tests were shown in 20% to 40% of various groups of patients with diabetes, with up to 20% having cardiovascular abnormalities and 12% having postural hypotension (58,59,60). However, although almost all patients with sensory neuropathy have an associated autonomic neuropathy, the autonomic abnormality is rarely symptomatic.
Many studies show a clear correlation between diabetic neuropathy and foot ulceration, with up to 80% of patients with foot ulcers having a clinically significant neuropathy (61,62,63,64). The positive predictive value of a clinically detectable neuropathy was shown in a prospective study, in which an increased vibration perception threshold was associated with a sevenfold increased risk of ulceration over 4 years (65). A more clinically useful examination, the Semmes-Weinstein nylon monofilament test, has also been shown to identify those at increased risk for ulceration (61,66,67,68). The inability to feel the 10-g filament greatly increases the patient’s risk of developing an ulcer. Undoubtedly, neuropathy plays a considerable role in the development of diabetic foot ulcers.
Sensory neuropathy is by far the major contributor to ulcer formation. Specifically, loss of pain sensation typically leads to repetitive trauma from walking, usually with poorly fitting shoes or from a foreign body in the patient’s shoe that the patient does not feel. This is compounded by loss of proprioception, which can lead to abnormal foot posture and amplified trauma from walking. Additionally, autonomic neuropathy can lead to sudomotor abnormalities, resulting in decreased sweating and dry skin that is prone to cracking. These breaks in the skin are portals for infection and further ulcer formation.
As autonomic neuropathy progresses, there is sympathetic denervation of the foot, resulting in loss of vasoconstrictor tone and peripheral vasodilation. As arteriovenous shunts open, there is, on average, a fivefold increase in blood flow to the skin, even in the absence of other signs of neuropathy (69,70,71,72). Clinically, the skin is deceptively warm and healthy appearing, with bounding pulses, marked venous distention, and an elevated venous Po2 secondary to shunting of blood (73). Increased capillary pressure may result in neuropathic edema that can further compromise tissue integrity (74). It has been suggested that shunting may bypass capillary beds, resulting in compromised flow of nutrients and oxygen to the bypassed tissues (75). However, it has been shown that nutritive capillary flow is not only normal but often increased (71). Blood flow to bone is also increased and may be responsible for the osteopenia of advanced disease, predisposing the patient to the development of Charcot neuroarthropathy (76).
Neuropathy alone is not sufficient to cause ulceration. Clearly trauma of some form is necessary. With time, loss of pain sensation in the foot can be compounded by structural abnormalities (34). Damage to motor nerves, specifically the tibial nerve, can lead to weakness and wasting of the intrinsic muscles of the foot. This leads to an imbalance between the flexor and extensor forces, deforming the shape of the foot itself. The metatarsal heads become more prominent, with unopposed dorsiflexion of the metatarsophalangeal joints; exaggerated interphalangeal joint plantarflexion results in clawing of the toes. The weight of the body is shifted onto a smaller surface, mainly the metatarsal heads and heel. In the early phases, the patient has a red, swollen foot with a clear sensory deficit on examination. As the neuropathy progresses, daily activities in the absence of adequate protective proprioceptive and nociceptive function lead to joint erosions, unrecognized fractures, and demineralization and devitalization of bones. Neuroarthropathy or Charcot joints develop as the foot collapses, the longitudinal arch flattens, and a rocker bottom deformity with weight bearing on the distal tarsal row develops, leaving the patient with an insensate misshapen foot (Fig. 68.2). Deformity becomes a constant source of abnormal pressure that in synergy with sensory dysfunction leads to ulceration. In one study, neuropathic feet with abnormally distributed pressures resulted in a 28% ulceration rate over 2.5 years as compared with no ulcers in neuropathic feet with normal pressures (77).
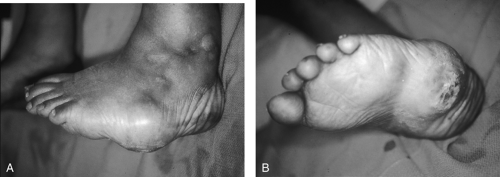 Figure 68.2. A, B: Charcot foot. Note ulcer forming over area of abnormally high pressure in this misshapen foot. (See also color plate.) |
Sites of high pressure loading, such as the metatarsal heads and heel, are prone to callus formation as exuberant keratin production is stimulated by high shear forces (Fig. 68.1B,C) (78). Callus buildup itself leads to further increases in pressure loading, which eventually results in foci of hemorrhage, liquefaction necrosis, and ulcer formation. Callus over a weight-bearing area is strongly predictive of ulcer formation, whereas callus removal decreases local pressures and the risk of ulceration (79,80). Clearly, the combination of neuropathy and altered pressure loading is a major risk factor for foot ulceration in the patient with diabetes. Deformity leads to altered pressure loading, which leads to callus formation and ultimately to ulceration. Repeated and continuous trauma disrupts the orderly sequence of cellular events necessary for wound healing and results in a nonhealing wound. Unchecked, these processes can result in a vicious cycle of further deformity, abnormal pressure loading, and progression of tissue breakdown.
The etiology of neuropathy in the patient with diabetes is complex and likely multifactorial. The ravages of neuropathy begin distally and are most likely to affect longer nerves first, such as those in the feet. This may be the result of the cumulative effect of multiple insults, both biochemical and vascular in nature, or increased capillary hydrostatic pressure, which is relatively unique to the feet and lower extremities (33,53). The association between hyperglycemia and worsening neuropathy is convincing and has been demonstrated by several groups (51,52,53,54). Furthermore, strict glucose control using continuous subcutaneous insulin infusion or pancreas transplantation can
slow or even halt the progression of neuropathic changes (45,81,82). The DCCT showed that intense glucose control led to a 69% reduction in subclinical neuropathy and a 57% reduction in clinically evident neuropathy (83). Some of the more convincing data regarding the pathogenesis of diabetic neuropathy implicate activation of the sorbitol pathway, the formation of advanced glycation end products, and increased oxidative stress (84,85,86,87,88,89,90,91,92,93). Animal models show that hyperglycemia can augment the sorbitol pathway in peripheral nerves. The products of this pathway can either directly or indirectly lead to nerve damage. Unfortunately, these animal data are not easily reproduced in human studies (94,95,96,97).
slow or even halt the progression of neuropathic changes (45,81,82). The DCCT showed that intense glucose control led to a 69% reduction in subclinical neuropathy and a 57% reduction in clinically evident neuropathy (83). Some of the more convincing data regarding the pathogenesis of diabetic neuropathy implicate activation of the sorbitol pathway, the formation of advanced glycation end products, and increased oxidative stress (84,85,86,87,88,89,90,91,92,93). Animal models show that hyperglycemia can augment the sorbitol pathway in peripheral nerves. The products of this pathway can either directly or indirectly lead to nerve damage. Unfortunately, these animal data are not easily reproduced in human studies (94,95,96,97).
Glucose uptake in peripheral nerves occurs independently of insulin. As a result, serum hyperglycemia leads to intracellular hyperglycemia and activation of the enzyme aldose reductase, stimulating the production of sorbitol (33,98). The concentrations of the intracellular osmolytes sorbitol, myoinositol, and taurine normally respond to changes in extracellular osmolality, buffering the intracellular environment (99,100,101). However, as hyperglycemia artificially drives the sorbitol pathway, intracellular accumulation of the osmolyte sorbitol leads to compensatory depletion of myoinositol and taurine (84,85,102,103). Initially, nerve damage was attributed to the intracellular accumulation of sorbitol and the resultant increased influx of water. This increased water content was shown in both diabetic animal and human nerves and was found to be reversible with aldose reductase inhibitors (104). However, the magnitude of the osmotic change was believed to be too small to account for significant peripheral neuropathy. Alternatively, the depletion of myoinositol, a significant component of phosphoinositide metabolism and a key determinant in the Na+-K+-ATPase that maintains nerve membrane potential, may become limiting for phosphoinositide signaling. Taurine depletion can exacerbate oxidative stress and lead to dysregulation of neuronal hyperexcitability and neurotransmitter release (85,102,105,106,107,108,109,110). The nerves of diabetic rats were found to have decreased myoinositol levels. The severity of myoinositol depletion correlated with decreased Na+-K+-ATPase activity and slowing of nerve conduction velocity. Furthermore, both defects were corrected with myoinositol supplementation or aldose reductase inhibitors (111,112,113,114,115,116). Although these data are intriguing, not all the evidence is supportive (117,118,119,120).
Activation of the sorbitol pathway consumes NADPH (reduced form of nicotinamide adenine dinucleotide phosphate), a cofactor necessary for the function of glutathione reductase and nitric oxide synthase activity, leading to depletion of both reduced glutathione and nitric oxide (92,121,122,123,124,125). Glutathione is a potent antioxidant and a critical element in the cellular defense against oxidative stress. Although direct evidence is lacking, oxidative tissue damage in diabetes has been attributed to NADPH consumption and the resultant depletion of reduced glutathione (121,126). Nitric oxide is a potent endothelium-derived mediator of vasodilation and neurotransmission (122,127). Aldose reductase-mediated depletion of NADPH can hinder nitric oxide synthase activity, which is NADPH dependent. Decreased nitric oxide production by the endothelial cells of the vasa nervorum can lead to limited vasodilation and impaired blood flow in the endoneurium of peripheral nerves, leading to local ischemia and neuronal dysfunction (92,98,121,123,124,125,128,129).
Activity of the sorbitol pathway can generate highly reactive sugar moieties that can glycate proteins, forming advanced glycation end products (AGE) (33,130). In general, high glucose concentrations can lead to the nonspecific glycation of both intracellular and extracellular proteins. Insulin-independent tissues such as peripheral nerves are particularly prone to hyperglycemia-induced glycation of intracellular proteins. Glycation of tubulin, a component of microtubules in nerves, can interfere with axonal transport, and AGE can nonspecifically consume nitric oxide, quenching its local vasodilatory effects and predisposing to neural ischemia (121,131). Aminoguanidine, an inhibitor of AGE formation, can reverse nerve conduction and blood flow abnormalities in diabetic rats (130,132,133). Finally, extracellular AGE are felt to play a role in the development of large-vessel atherosclerosis and ischemia in the patient with diabetes. AGE-induced abnormalities in the extracellular matrix have been shown to alter the structure and function of the vasculature, decreasing elasticity in large vessels and increasing permeability across the carotid artery in rats (130). By blocking the cytostatic effect of nitric oxide on vascular smooth muscle, accumulation of AGE is thought to contribute to the premature and accelerated evolution of atherosclerosis and large-vessel occlusive disease in the diabetic patient (130).
ISCHEMIA
Ischemia and its complications are responsible for some of the most devastating ravages of diabetes mellitus. Patients with
diabetes are at an increased risk of developing atherosclerotic disease of both large and small vessels and do so at an earlier age and at an accelerated rate (134,135). More than 80% have vascular disease 20 years after diagnosis of their diabetes, and 75% of patients with diabetes die of vascular disease or its complications (136). Ischemia clearly plays a role in diabetic foot ulcers as well. Tissue hypoxia as measured by a reduction in dorsal foot transcutaneous oxygen tension has been shown to be an independent predictor of foot ulceration in the diabetic patient (137). However, purely ischemic ulcers are relatively uncommon, representing only 10% to 15% of diabetic foot ulcers (63,138,139). Most of these ulcers are neuropathic or neuroischemic in nature (54,75,134,140,141). Certainly, the metabolic requirements of a wound are greater than those of uninjured tissues (142). Therefore, a diabetic foot with adequate tissue perfusion may become ischemic once it is wounded if it is unable to augment blood flow to the injured tissues. Vascular occlusive disease in combination with an abnormal autonomic response secondary to neuropathy may limit the ability to augment flow. This creates the vicious cycle of a wound leading to ischemia, leading to a larger wound, which, if uninterrupted, often leads to amputation.
diabetes are at an increased risk of developing atherosclerotic disease of both large and small vessels and do so at an earlier age and at an accelerated rate (134,135). More than 80% have vascular disease 20 years after diagnosis of their diabetes, and 75% of patients with diabetes die of vascular disease or its complications (136). Ischemia clearly plays a role in diabetic foot ulcers as well. Tissue hypoxia as measured by a reduction in dorsal foot transcutaneous oxygen tension has been shown to be an independent predictor of foot ulceration in the diabetic patient (137). However, purely ischemic ulcers are relatively uncommon, representing only 10% to 15% of diabetic foot ulcers (63,138,139). Most of these ulcers are neuropathic or neuroischemic in nature (54,75,134,140,141). Certainly, the metabolic requirements of a wound are greater than those of uninjured tissues (142). Therefore, a diabetic foot with adequate tissue perfusion may become ischemic once it is wounded if it is unable to augment blood flow to the injured tissues. Vascular occlusive disease in combination with an abnormal autonomic response secondary to neuropathy may limit the ability to augment flow. This creates the vicious cycle of a wound leading to ischemia, leading to a larger wound, which, if uninterrupted, often leads to amputation.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


