CYTOMEGALOVIRUS
PAUL D. GRIFFITHS
HISTORY
In the early years of the twentieth century, histopathologists studying stillborn infants identified the characteristic intranuclear inclusions in fatal cases of what is now termed cytomegalic inclusion disease (1). At first, the inclusion bodies were attributed to a new protozoal infection (2). However, in 1921 Goodpasture and Talbot (3) remarked on the similarity between these strange bodies and the intranuclear inclusions produced in the skin lesions of varicella (4). The viral etiology of the inclusions was first proposed by von Glahn and Pappenheimer (5) in 1925. The following year, Cole and Kuttner (6) were able to transmit the guinea pig form of cytomegalic inclusion disease from salivary gland material passed through a Berkefeld N filter. Later, three laboratories simultaneously reported the isolation of the causative human virus in cell cultures: Smith (7) in 1956 from salivary glands, Rowe et al. (8) in 1956 from adenoid tissue, and Weller et al. (9) in 1957 from a liver biopsy specimen. In 1960, Weller et al. (10) named the virus “cytomegalovirus” (CMV).
Since a characteristic feature of cytomegalic inclusion disease is global mental retardation, scientists have known for more than 50 years the basic nature of CMV and its tropism for cells of the central nervous system (CNS). However, whether this tropism resulted from the immature phenotype of cells dividing to form the fetal CNS or whether CMV could also infect terminally differentiated neural cells of adult origin was not known. Although individual cases of CMV encephalitis have been described in patients who were immunocompromised but not infected with human immunodeficiency virus (HIV), such cases are still the exception rather than the rule. For example, CMV encephalitis was described in a renal transplant recipient treated with augmented immunosuppression for graft rejection (11) and in a patient with Hodgkin disease who became demented and died of pneumonia (12). The acquired immunodeficiency syndrome (AIDS) epidemic changed this perspective; CMV infection was found so often at autopsy before the advent of highly active antiretroviral therapy (HAART) that it was the most common opportunistic infection of the CNS in patients with AIDS (13). Both CMV infection and disease, including of the CNS, have decreased dramatically in countries where HAART is readily available. This clinical benefit is consistent with the possibility that CMV infection of the CNS was a major and often unrecognized contributor to CNS disease. However, the clinical course of patients with AIDS is so complex that it is not certain how much CNS disease could be attributed to the presence of CMV as opposed to HIV, which is also neurotropic and controlled by HAART. Interestingly, CMV encephalopathy can predominate as the cause of death in a child who acquires both CMV and HIV from its mother (14). The aim of this chapter is to review what is known and how much remains to be defined about CMV infection of the CNS.
INFECTIOUS AGENT
CMV has the largest genome (229 kb) of the viruses known to infect humans. More than 200 potential open reading frames were identified in strain Ad169 (15), with an additional 22 in wild type strains (16). Propagation in fibroblasts selects for this and other genetic changes so viruses are now maintained in the laboratory as bacterial artificial chromosomes with approximately 165 protein-coding genes in wild type strains (17). In addition, the genome encodes approximately 10 micro-RNAs. Recent results with ribosome profiling suggest that the genetics of this virus may be even more complex than described so far (18). Salient features of the molecular biology of the virus are summarized later in this chapter, with a focus on genes important for immune control or which are the targets of antiviral chemotherapy; for extensive details, readers are referred elsewhere (19). By international agreement, proteins encoded by the virus are described according to their map position (20) (Fig. 11.1). For example, gpUL55, the glycoprotein known as glycoprotein B, originates from the fifty-fifth open reading frame of the unique long region, while ppUL82 is a tegument phosphoprotein transactivator that maps to the eighty-second open reading frame of the unique long region.
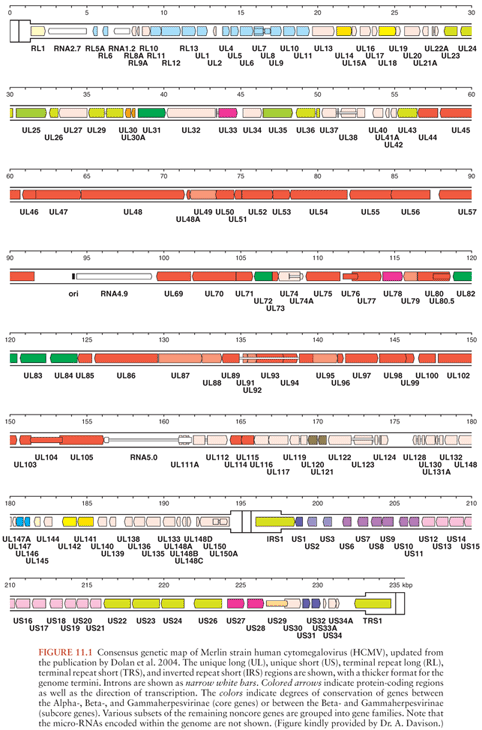
CMV encodes several transactivators. The major immediate-early region maps to UL122/123. Alternative splicing produces four major proteins, one of which interacts directly with TATA-binding protein and downregulates its own promoter. This region contains upstream enhancers, is a strong regulatory region, and is a major target of cell-mediated immunity. pUL69 is a transactivator found in the virion. It synergizes with ppUL82 to activate the major immediate-early promoter. These two proteins together can thus be thought of as being functionally equivalent to the α-transinducing factor of herpes simplex virus (HSV).
A total of 11 genes are required for CMV replication (21). These proteins together provide in trans the functions required to allow replication of the lytic origin of replication (22). Some of the functions are directly analogous to those required by HSV, for example, DNA polymerase (UL54) and its accessory protein (UL44), although an equivalent to the origin-binding protein has not been identified.
Structural proteins of the virus include the major capsid protein, UL86, and the minor capsid protein, UL46. The assemblin gene complex (UL80a) includes a protein important for packaging DNA as well as a protease. Several phosphoproteins are found in the tegument (UL32, UL83, UL82, UL99). ppUL83, known as the lower matrix protein, is the major antigen detected in circulating leukocytes (23) and is the second major target of the cell-mediated immune response (24).
Major envelope proteins exist on the plasma membrane as multimolecular complexes and are major targets for humoral immune responses. Glycoprotein complex I consists of a homodimer of glycoprotein B (gB; gp UL55). Glycoprotein complex II consists of gM (gp UL100) and gN (gp UL73). Glycoprotein complex III consists of gH (gp UL75), gL (gp UL115), and gO (gp UL74) in fibroblasts but gH, gL, and three proteins (pUL128, pUL130, pUL131A) encoded within the region of the genome which is deleted upon passage in fibroblasts. This five-member complex of proteins mediates viral entry into epithelial and endothelial cell lines (25).
CMV encodes a series of genes that interfere with immune recognition of virus-infected cells. The product of US6 blocks the transporter associated with antigen presentation, which normally takes peptides from the cytosol into the lumen of the endoplasmic reticulum. US3 binds to human leukocyte antigen (HLA) heavy chains and retains them in the endoplasmic reticulum. Genes US2 and US11 take HLA complexes from the endoplasmic reticulum and export them back into the cytosol for degradation in the proteasome. All of these effects combine to decrease HLA display at the plasma membrane and thus allow the virus to escape from cytotoxic T cells. However, because HLA molecules also provide non–antigen-specific signals to natural killer (NK) cells and macrophages, their absence could trigger destruction of the virus-infected cell via these innate immune effectors. To avoid this, CMV has additional genes: UL18 to provide a negative signal to macrophages and UL40 to provide a negative signal to NK cells. In addition, the proteins encoded by UL16, UL141, and UL142 block the recognition of a positive signal for NK cells. In addition, a viral micro-RNA interferes with recognition of another of these host defense signals. In combination, the function of these genes allows CMV to persist in the host despite strong humoral and cell-mediated immune responses.
CMV encodes four sets of proteins that are predicted G protein–coupled receptors (US27 and US28; UL33; UL78). UL97 is a protein kinase homolog responsible for phosphorylating ganciclovir and acyclovir (26–28).
EPIDEMIOLOGY
Humans are the only reservoir for CMV (29). Infection is endemic worldwide and has no seasonal variation. The prevalence of CMV infection increases directly with age, with significant variations according to geographic, ethnic, and socioeconomic backgrounds. Antibodies of immunoglobulin G (IgG) class against CMV can be found in approximately 60% of adults in developed countries, and virtually 100% of adults in developing countries (30). Within developed countries, acquisition of CMV infection is increased in poor socioeconomic environments and by sexual contact. The presence of IgG antibodies indicates that infection has occurred sometime in the past and that the virus should be presumed to be latent. The sites of latency of CMV are not known, although myeloid dendritic cells are clearly one of them, with reactivation of virus linked to cellular differentiation (31–33). CMV can be transmitted iatrogenically by all organ allografts and at autopsy can be found in most tissues of the body. Thus, whether CMV has a single site of latency or whether silent infection can become established in multiple tissues is debatable.
Transmission of infection from one individual to another requires direct contact. Sources of virus include saliva, blood, vaginal secretions, semen, and breast milk (34–36). Salivary excretion of virus, particularly in association with mouthing of toys, can lead to baby-to-baby transmission in the child care setting (37–39). Susceptible children can thus acquire CMV in the day care environment and transmit infection to susceptible family members in the home. Transmission of CMV from an infant to his or her seronegative mother—as well as from an infant to pregnant caretakers, with subsequent transmission to the fetus—has been documented (40,41). Overall, in the United States, contact with young children is a much more important source of CMV for mothers than is sexual exposure (42).
Prospective studies in the United States and Europe have established that pregnancy itself does not increase the risk of acquiring CMV infection (43–50). Primary CMV infections during pregnancy are generally asymptomatic (43,45–47,49–51). Among susceptible women, the risk of seroconversion during pregnancy averages 1.7% (52). In primary CMV infection, an innate barrier prevents in utero transmission; thus, primary maternal CMV infection leads to transmission in only about 32% of fetuses (53). Moreover, only 12.7% of infected infants have clinical manifestations at birth (from very mild to severe), and the risk of subsequent sequelae averages 13.5% (54). Although the rate of transmission of virus in utero appears lower, the risk of neonatal disease with attendant sequelae appears to be higher when the mother has acquired CMV infection during the first trimester of pregnancy (55).
Maternal immunity to CMV neither prevents virus reactivation nor controls the systemic spread of virus that can produce congenital infection (44,48,56–60). Studies that use restriction endonuclease analysis of viral DNA to examine the genetic relatedness between CMV strains isolated repeatedly from mother and offspring indicate reactivation of an identical latent virus rather than reinfection with an exogenous one (61). In other cases, there is evidence for exogenous reinfection with a different strain (62,63). In a highly seropositive (82% of persons) urban population of low socioeconomic background in Alabama, recurrent CMV infection produced a 1.6% (13/835 infants) rate of congenital infection, thus indicating that most intrauterine infections in this population result from this phenomenon (49). In contrast, the infants of immune middle-class women with a seropositivity rate of 55% had a lower rate of congenital infection (0.19%, i.e., 10/5,242). This rate is not significantly different from that observed in Great Britain and Sweden (44,48,57). Overall, the mothers of only 25% of babies born with congenital CMV infection in the United States had primary infection during pregnancy (64). Although maternal immunity is imperfect, congenital infections that result from recurrent infection are less likely to produce fetal damage than those resulting from primary infections (49). However, the larger number of seropositive women in a community may provide a number of infected neonates equal to those born to seroconverting mothers (65).
During pregnancy and in the immediate postpartum period, CMV can be shed at variable rates from one or more sites (49). Rates of excretion are higher in younger, nonwhite women from lower socioeconomic backgrounds. Most women who excrete virus during pregnancy do so as a result of recurrent infections (reactivation or reinfection) (61). There is little correlation between CMV excretion from the cervix or in urine during pregnancy and the subsequent birth of a congenitally infected infant (47,50,66), but the presence of CMV in the maternal genital tract at delivery and in the breast milk after delivery is strongly associated with intrapartum/postpartum transmission of infection to the infant (34,36). In full-term and otherwise healthy infants, perinatal infection has little clinical importance except for a small number of cases of interstitial pneumonia (36,67,68). However, such children are a major source of infection for other children in the day care environment (69,70) and represent an occupational risk to child care personnel, particularly women of childbearing age (39,71–73).
If a patient who is CMV IgG-seropositive becomes immunosuppressed, CMV reactivation is common, being found typically in 50% of transplant recipients at some time after transplantation. The donor organ can also transmit CMV to seronegative individuals (74) or to seropositive individuals (75), showing that preexisting immunity to CMV does not provide protection against infection. However, preexisting immunity does provide moderate protection against CMV replication to high levels and disease (76). Primary CMV infection can also be acquired from blood transfusion (77).
In patients who are HIV positive, the same principles apply: CMV infection becomes increasingly common as the immunocompromised state progresses, although formal studies of natural history have not been performed. However, CMV disease, typically retinitis, clearly is uncommon before the CD4 count declines to 100 × 106/L. It becomes increasingly common as such patients are followed, with typically 10% of patients developing CMV disease within 2 years of follow-up (78). Retinitis is associated with CMV encephalitis (79). By itself, it represents approximately 85% of CMV disease in patients with AIDS, with gastrointestinal tract infection representing approximately 10% and CMV CNS disorders only 1% of total disease. Nevertheless, presentations in the CNS (and peripheral nervous system) are being increasingly recognized, as described later in this chapter.
ETIOLOGY
In cases of congenital CMV, one can be certain that the CNS disease is attributable to CMV, as no other infectious agent is present and the syndrome is characteristic. The same is probably true in the case of a female preterm infant born to an HIV-positive mother who acquired congenital CMV and HIV infection vertically. At 5 months, the child developed AIDS and died at age 11 months of a rapidly progressive diffuse encephalopathy. At autopsy, CMV encephalitis was seen, with occasional inclusion bodies, yet there was no evidence for HIV infection of the brain (14).
However, the same clear-cut picture is not seen in adult patients with AIDS, who typically have evidence for HIV involvement of the CNS, with or without CMV infection (80). A case definition for CNS conditions attributable to HIV has been provided (81). However, it is not possible to exclude an HIV contribution to CNS symptomatology in patients with CMV. Indeed, in vitro there are multiple ways in which CMV and HIV could interact so that each could enhance the replication of the other; this has been reviewed by Griffiths (82). CMV infection can also downregulate HIV (83). Other opportunistic infections of the CNS are usually spatially distinct from CMV, with the exception of HSV type 1 (HSV-1); superimposed CMV/HSV-1 infection has been described (84,85). Interestingly, superimposed CMV/HSV-1 infection of the brain has also been described in a patient who does not have AIDS (86).
Cases have been described in which CMV appears to be the cause of CNS dysfunction (87–89). Until more is known about the pathogenesis of CMV infection of the CNS, however, it is not possible to guarantee that CMV infection is the sole cause of the CNS dysfunction in an individual patient. Nevertheless, populations of patients with AIDS can be examined to discern patterns of clinical syndromes, with the strong presumption that CMV is a candidate etiologic agent.
Animal experiments are not particularly helpful, because human CMV is strongly species specific and replicates only in humans. Animal CMVs, such as murine or guinea pig CMV, can be studied; although they are different viruses, they do have the advantage of occupying the same ecologic niche in their species as human CMV does for humans. For example, studies of the direct intracerebral inoculation of murine CMV into mice of different ages show that the virus has a predilection to infect developing brain cells (90).
PATHOGENESIS
Results of preliminary studies indicate that the virulence of congenital CMV infection is not strain dependent (61). Although only a few infants have been studied, viruses isolated from congenitally or perinatally infected siblings were genetically identical. In two of the pairs of siblings assessed, the first-born baby was severely affected, whereas the second-born infant was only subclinically infected (60). Mechanisms that possibly explain some of the CNS damage associated with CMV infection include productive CMV replication leading to destruction of individual cells (lytic infection) and indirect damage mediated by action of components of the immune system (immunopathology).
Lytic Infections
Continuous viral replication in affected organs could explain why some infants are severely affected and others remain free of symptoms. Longitudinal studies have demonstrated that excretion of CMV into urine and saliva persists for years. Chronic viral replication probably also occurs at other anatomic sites that are less accessible to virologic examinations (e.g., middle ear, brain).
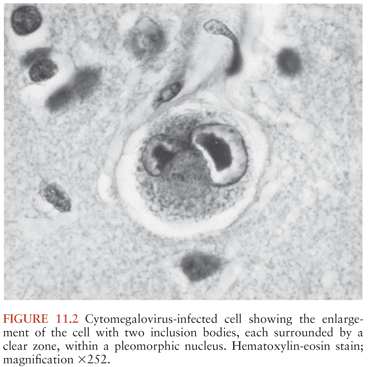
The histopathologic hallmark of CMV CNS infection is the characteristic intranuclear inclusion body (Fig. 11.2). This indicates productive infection of the cell and represents a major site of virus formation. Such cells are likely committed to die, so CNS dysfunction could result from loss of their function. Inclusion-bearing cells were seen in six fetuses with polymerase chain reaction (PCR)–positive amniotic fluids that were terminated at 19 to 35 weeks because of CNS lesions seen on ultrasound examinations (91). Meningoencephalomyelitis and micronodular encephalitis were seen in all, consistent with bloodborne dissemination of virus. Diffuse periventricular microglial nodules were present. Inclusions were seen in the stria vascularis, Reissner membrane, and the organ of Corti as well as in epithelial cells of the saccule and utricule. The authors suggested that CMV may gain access to the inner ear via the highly vascularized stria vascularis and damage the ability of this structure to maintain potassium homeostasis, dissipate electric potentials, and support recirculation of potassium within the perilymph.
Immunopathology
Studies of CMV polypeptides immunoprecipitated by IgG antibodies indicate that symptomatically infected infants had a delay (until 12 months of age) in the appearance of precipitating antibodies (92). However, when a humoral immune response develops, antibodies to viral polypeptides are precipitated in greater numbers and are maintained for longer periods. This sustained humoral immune response to congenital CMV infection occurs in the presence of persistent viral replication, thereby creating the potential for the formation of immune complexes (93). During the first year of life, immune complexes circulate in a large proportion of infants with congenital infection. The molecular weight of these immune complexes is higher in symptomatic infants than in asymptomatic infants. In a few fatal symptomatic cases, deposition of immune complexes in renal glomeruli has been demonstrated. Another factor in disease pathogenesis is vasculitis, which may occur in utero or after birth. Infants with congenital CMV infection who die soon after birth usually have disseminated intravascular coagulopathy.
CMV pneumonitis in transplant recipients may be immunopathologically mediated (94). Thus, the presence of the virus is required for the syndrome, but the clinical disease may be caused by an abnormal, enhanced, cell-mediated immune response. Immunopathology presumably also explains the immune recovery vitreitis, which follows successful treatment of CMV retinitis in patients with AIDS. Similarly, it has been suggested that the demyelinating component of the disease in the peripheral nervous system could be immunopathologically mediated (95). It is unlikely that an identical scenario pertains in the CNS of patients with AIDS, because neurologic symptoms usually occur at profoundly low CD4 cell counts. However, there are distinct possibilities that host cytokines, released as a result of CMV infection, stimulated by CMV, or even encoded by CMV, could mediate extensive neural damage. For example, CMV activates tumor necrosis factor-α and interleukin-6 (IL-6), both of which may induce HIV expression in macrophages (96). CMV replication is also enhanced by IL-8. CMV infection induces production of messenger RNA (mRNA) of the IL-8 receptor, while IL-8 increases CMV replication approximately fourfold (97). pUS28 has 33% amino acid homology with the IL-8 receptor (97). Because IL-8 stimulates production of polymorphonuclear leukocytes, this is a plausible explanation for the increased level of polymorphonuclear leukocytes seen in the CNS in association with CMV, particularly polyradiculopathy. The CMV genome encodes an α-chemokine (UL146) and an IL-10 homolog (UL111A) whose function remains undefined in vivo.
PATHOLOGY
CMV reaches the brain through viremic spread. In patients with AIDS, viremia has been reported to have both a cellular and a plasma component (98). In healthy individuals, CMV can be found by PCR in monocytes (31). In transplant recipients, ppUL83 (pp65) antigen has been demonstrated in monocytes, polymorphonuclear cells, or endothelial cells (23). Which, if any, of these cells are important for disseminating CMV to the brain of patients with AIDS is not clear, but there is evidence that microglia are derived from the peripheral blood monocyte pool (99,100). Regardless, there is evidence that CMV can spread through the brain by two routes.
The first route involves infection of the endothelial cells of the brain, with spread to the contiguous astrocytes in foot processes and their cognate neurons. This route of inoculation is mirrored by the following histologic findings: (a) infection of endothelial cells in the brain, which may sometimes be associated with vascular thrombosis. Isolated endothelial cells containing inclusion bodies may also be seen in the spinal cord in association with a myelitis; (b) multiple foci of glial nodular encephalitis. The glial nodules consist of multiple microglial cells and some astrocytes. In some cases, the microglial response surrounds an inclusion-bearing cell. Other inclusion body cells may be missed in the plane of section. In the series by Vinters et al. (85), CMV could not be found in every case of microglial nodular encephalitis identified. Infected cells can likely be targets for microglial attack before reaching the stage of bearing an inclusion body, because staining with monoclonal antibodies or the use of in situ hybridization reveals many more cells to be CMV infected (101,102).
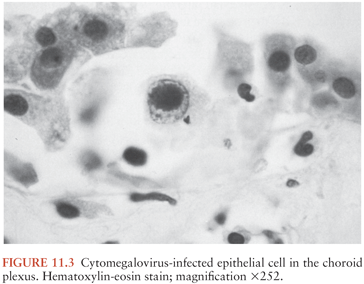
The second route of viremic spread is through the cerebrospinal fluid (CSF) following viral replication in infected epithelial cells of the choroid plexus (Fig. 11.3). Secondary seeding of the ependymal surfaces and internal spread produce necroses of periventricular white matter (102). This condition, necrotizing ventricular encephalitis, may be extensive. Ependymal surfaces are replaced with CMV-infected cells, infiltrating macrophages, inflammatory exudate, and necrotic cells with or without hemorrhage (85,101).
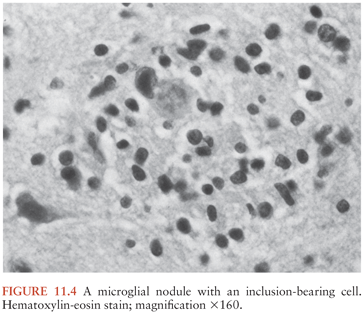
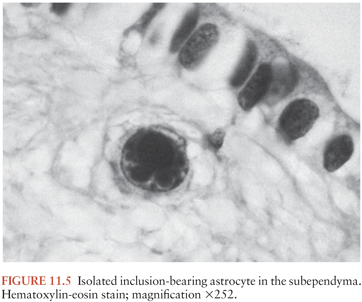
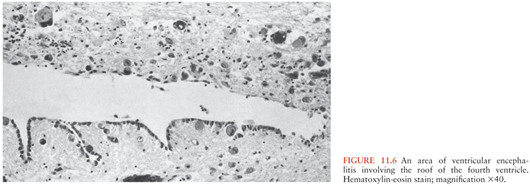
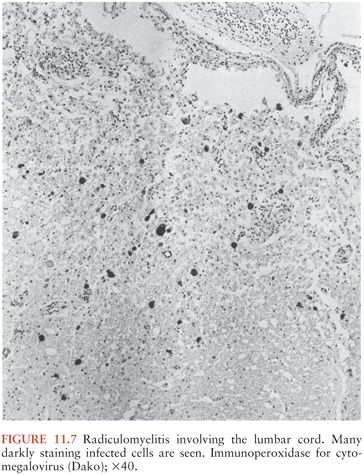
The histologic changes may be classified into one of five major groups: (a) nodular encephalitis, in which widely disseminated microglial nodules, only a minority of which may contain inclusion-bearing cells (Fig. 11.4), are seen disseminated over a wide area (103); (b) isolated inclusion-bearing cells, which may be astrocytes (Fig. 11.5), neurons, or macrophages (104); (c) focal parenchymal necroses in an otherwise normal neuropil, whose lesions are characterized by accumulations of macrophages with axonal destruction and occasional inclusion-bearing cells (105); (d) ventricular encephalitis, characterized by inflammation and necrosis involving the ependyma and subependymal glia (Fig. 11.6), often over a wide area of the ventricular lining (106); and (e) radiculomyelitis, in which inflammatory changes, often with a marked infiltrate of polymorphonuclear neutrophils, are seen involving the cord (Fig. 11.7), spinal roots, and dorsal root ganglia. To some extent, the histologic changes mirror the portal of entry of the virus into the CNS and its dissemination throughout the brain and spinal cord, as discussed earlier.
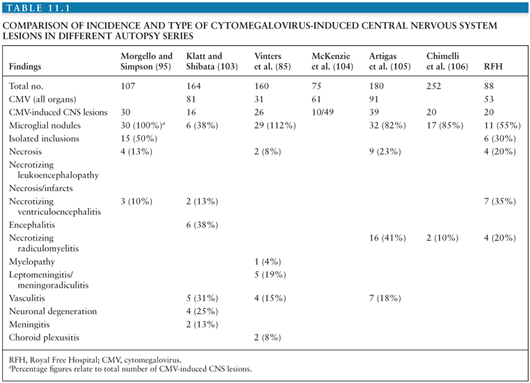
The incidence of such findings in seven separate AIDS autopsy series is shown in Table 11.1. The proportion of patients in whom CMV infection of the CNS was documented varied from 7.9% to 28%. In total, 161 (15.7%) of 1,026 patients in these studies had CMV involvement of the CNS.
An increasing number of viruses are being described that produce similar clinical and pathologic changes in patients with AIDS (107). Indeed, the clinical and pathologic features may reflect more the route of infection and the state of the immune system than the nature of the virus.
CLINICAL MANIFESTATIONS
Clinical Presentation of Congenital Infection
Symptomatic Infection
Disease in the symptomatic newborn is characterized by hepatomegaly, splenomegaly, microcephaly, direct hyperbilirubinemia, petechiae, and thrombocytopenia (108). Intracranial calcifications and retinitis are demonstrable with regularity. Intrauterine growth retardation is often present. Diabetes insipidus has been reported in a few infants with congenital infection (109).
The mortality among congenitally infected infants may be attributed to hepatic dysfunction, bleeding, or secondary bacterial infection (Table 11.2). Death after the first month of life is usually the consequence of hepatic dysfunction; however, beyond the first year of life, death is usually the result of complications in the neurologically handicapped child secondary to malnutrition, aspiration pneumonia, or overwhelming infection.
Related posts:
 Neurologic Manifestations of Varicella and Herpes Zoster
Neurologic Manifestations of Varicella and Herpes Zoster
 Meningitis and Encephalitis Caused by Mumps Virus
Meningitis and Encephalitis Caused by Mumps Virus
 Human Immunodeficiency Virus
Human Immunodeficiency Virus
 Vaccines for Viral Diseases with Significant Central Nervous System Manifestations
Vaccines for Viral Diseases with Significant Central Nervous System Manifestations
 Subdural Empyema and Suppurative Intracranial Phlebitis
Subdural Empyema and Suppurative Intracranial Phlebitis
 Rickettsioses, Anaplasmoses, and Q Fever
Rickettsioses, Anaplasmoses, and Q Fever
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


