FIGURE 27.1 Validated genes mutated or deregulated in melanoma progression. Progression of normal melanocytes to metastatic melanoma is diagrammed with associated genetic events as a linear process, although in a majority of the cases, melanomas may not arise from a pre-existing nevus. The degree to which the association of each gene has been experimentally validated is shown in Table 27.1.
TABLE 27.1
SUMMARY OF VALIDATED GENES INVOLVED IN MELANOMA GENESIS AND PROGRESSION
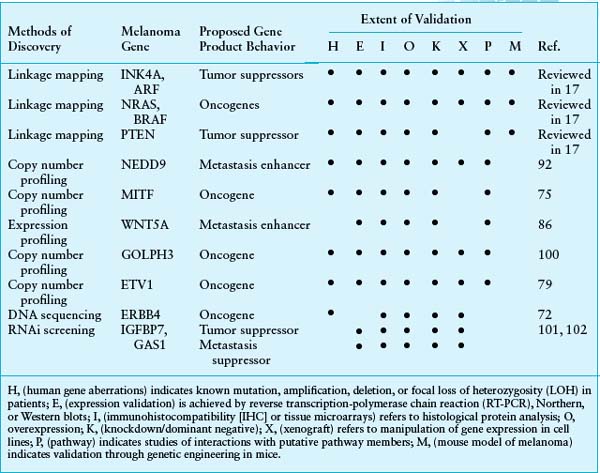
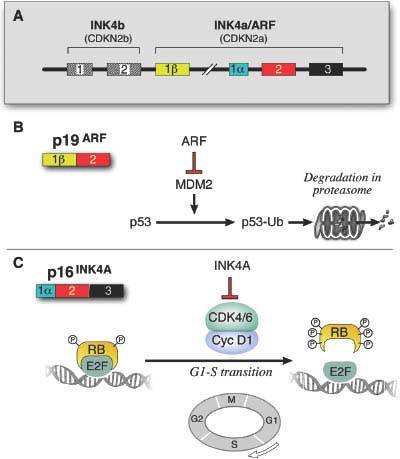
FIGURE 27.2 The unusual genomic structure and products of the CDKN2A locus. A: INK4A and ARF (p14ARF in human and p19ARF in mouse) initiate in different first exons and share the coding exon 2, but in an alterative reading frame, thus encoding two proteins with no amino acid similarity. The involvement of the neighboring and related CDKN2B locus in the pathology of melanoma is unclear, although it is often deleted in conjunction with CDKN2A. (From ref. 3.) B: ARF participates in the p53 pathway by binding to and sequestering the p53 antagonist, MDM2. The loss of ARF therefore results in the net degradation of p53 by MDM2-mediated ubiquitination. p16INK4A inhibits the action of the CDK4/CDK6-cyclin D1 complex in the retinoblastoma (RB) pathway. The loss of p16INK4A results in the phosphorylation and inactivation of pRB leading to its uncoupling from the transcription factor E2F, allowing for transcription of E2F target genes that are required for cell cycle progression from G1 to S phase.
CDK4
CDK4 is a direct target of inhibition by p16INK4A (Fig. 27.2) and is a primary regulator of RB activation. Rare germline mutations of CDK4 that render the protein insensitive to inhibition by INK4A (e.g., Arg24Cys) have been identified in a melanoma-prone kindred.9 These tumors retain wild type INK4A function, suggesting that INK4A is epistatic to CDK4 and that RB pathway deregulation is central to melanoma genesis. Somatic focal amplifications of CDK4 are also observed (albeit rarely) in sporadic melanomas.10 Carcinogen treatment induced melanomas in the animals without somatic Ink4a inactivation, similar to the mutual exclusivity observed in familial melanoma.11
RB1
Germline mutations in RB1 confer predisposition to melanoma in patients who have survived bilateral retinoblastoma.12 These melanomas exhibit loss of heterozygosity (LOH) of the remaining wild type RB1 allele. In such patients, estimates of increased lifetime risk of melanoma range from 4- to 80-fold. Interestingly, the RB1 gene locus is frequently deleted in primary cutaneous melanomas,13 and RB1 may be subject to genomic rearrangement in rare instances.14
THE p53 PATHWAY
The p53 pathway is critical for maintenance of the normal genome by regulating a multiplicity of mechanisms, including cell cycle checkpoints, DNA damage repair activation, and the appropriate induction of apoptosis. Mutations in the TP53 gene occur in over 50% of all tumors. Although the TP53 locus is rarely mutated in human melanomas,15 this region appears to undergo copy neutral LOH at enhanced frequency, at least in advanced tumors.4 Furthermore, loss of p53 in mice cooperates with activated H-Ras to induce melanomas.16 Thus, while TP53 is rarely deleted in human melanomas, inactivation of its pathway appears critical for melanoma genesis.
ARF
ARF-specific insertions, deletions, and splice donor mutations have been described in human melanomas.17 However, it remains ambiguous whether the genetic disruption of ARF alone is sufficient for tumorigenesis. In mouse models, Arf-specific deletion in conjunction with activated H-RAS leads to a similar melanoma phenotype as the Ink4a-specific deletion mouse mentioned above8; however, Arf-mutant melanomas were enriched for mutations in the RB pathway. Notably, upon UV radiation these mice developed focal amplifications at the Cdk6 locus,18 which encodes an orthologue to CDK4. Furthermore, p53 heterozygous mouse melanomas retain Arf, demonstrating their epistatic relationship.16
THE MAP KINASE PATHWAY
Extensive genetic and mechanistic studies have unearthed a prevalence of activating mitogen-activated protein kinase (MAPK) pathway mutations across many tumor types. The focal point of MAPK activation is the ERK1/2 kinases, which mediate the transcription of many genes governing cell proliferation and survival (Fig. 27.3). In nontransformed cells, key MAPK effectors have also been shown to regulate differentiation and senescence.
THE RAS FAMILY: H-, N-, AND K-RAS
Increasing evidence shows that the three different members of the classical RAS oncogene family exert functionally separable roles. N-RAS is the most frequently targeted in melanoma (33% of primary and 26% of metastatic samples19), followed by H-RAS (mainly in Spitz nevi).20 Despite their high incidence in other cancer types, K-RAS mutations are rarely observed in melanocytic lesions.21,22 Interestingly, although N-RAS mutations are found in 54% of congenital nevi, they are rare in dysplastic nevi,23 implying a distinct evolutionary path from dysplastic nevi to melanoma.
In mouse models, overexpression of activated H-RAS or N-RAS on an Ink4a/Arf-null background results in spontaneous melanoma formation.6,24 However, while H-RAS-induced melanomas rarely, if ever, metastasize, N-RAS tumors frequently metastasize to draining lymph nodes and distal organs, in line with the apparent selection for N-RAS over H-RAS mutations in human melanomas. Knockdown of NRAS in human melanoma cell lines inhibits their viability, indicating dependency on this oncogene for tumorigenicity.28 Furthermore, shutting off transgene expression in an inducible H-RAS model caused regression of melanomas that arose following transgene induction, thereby confirming the RAS oncogene dependency in these tumors.26
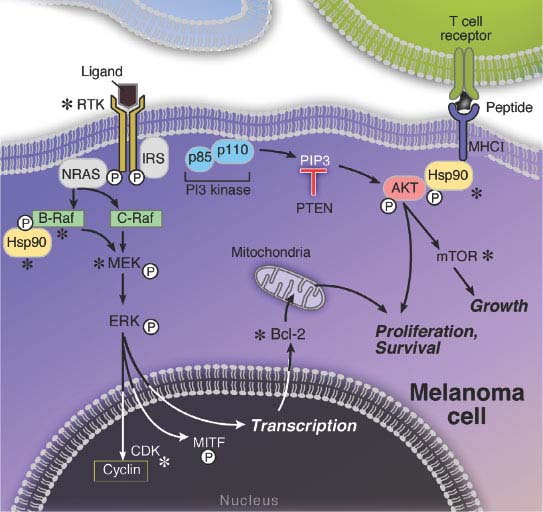
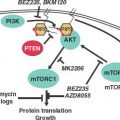
FIGURE 27.3 The melanoma signaling cascades mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K). The MAPK pathway is hyperactivated in melanomas, mainly due to activating mutations in either the NRAS or BRAF genes. The sequential phosphorylations of the downstream MEK and ERK proteins ultimately results in the activation of a number of transcription factors, including MITF, which induce cell proliferation and survival. The PI3K pathway is mainly hyperactivated by loss of the PTEN tumor suppressor, which results in the phosphorylation and activation of the survival gene AKT and subsequent stimulation of the mitogenic mammalian target of rapamycin (mTOR) pathway. The relative independence of the MAPK and PI3K pathways is supported by the apparent need for mutations that affect both.
BRAF
Somatic, activating BRAF mutations occur at high frequency in melanoma. These mutations are dominated by a single T→A transversion, resulting in a valine to glutamate amino acid substitution (V600E). Although the T→A transversion is not classically associated with UV-induced damage, BRAF mutations appear to be more common in melanomas arising at sites with intermittent exposure to UV.13,27 On the other hand, melanomas from chronically sun-damaged skin are typically wild type for BRAF.13 BRAF mutations may be an early preneoplastic event, given their high incidence in benign and dysplastic nevi (Fig. 27.1).28
BRAF is an immediate downstream target of RAS (Fig. 27.3) in the MAPK pathway. The BRAF(V600E) mutation confers more than 500-fold induction of kinase activity in vitro; this dramatic effect may explain why the V600E mutation dominates in BRAF-mutant melanoma. BRAF(V600E) mutations are often observed in conjunction with PTEN loss (see below), implying that dual modulation of MAPK and phosphatidylinositol 3-kinase (PI3K) pathways may promote a fully transformed phenotype.
Extensive data suggest that wild type BRAF operates on a senescence pathway in benign human nevi. Transgenic expression of BRAF(V600E) targeted to melanocytes in zebrafish produced benign nevuslike lesions, whereas invasive melanomas were produced (after extended latency) when crossed into p53 deficient zebrafish.29 Inducible expression of BRAF(V600E) alone in murine melanocytes resulted in excessive skin pigmentation and the appearance of nevi containing hallmarks of senescence.30 Human congenital nevi with activating BRAF mutations were shown to express senescence-associated acidic β-galactosidase (SA-β-Gal), the classical senescence-associated marker.2 This implied that activated BRAF alone is insufficient to induce tumor progression beyond the nevus stage in patients (Fig. 27.1). Interestingly, immunohistochemical staining of nevoid tissues found heterogeneous patterns of INK4A that only partially overlapped with SA-β-Gal, suggesting the presence of INK4A-independent pathway(s) operative in oncogene-induced senescence.
The high prevalence of activating BRAF and NRAS mutations implied that the MAPK cascade might confer a “drugable” melanoma tumor dependency when such mutations were present. This notion was supported by several lines of preclinical evidence, particularly in BRAF(V600E) melanomas. For example, constitutive ERK activation in BRAFV600E cells was required for their continued proliferation.31–33 RNAi knockdown of BRAF in this genetic context inhibited ERK activation, blunted cell growth, and in some cases induced apoptosis in vitro.31,32 BRAF silencing also suppressed anchorage-independent growth and tumor formation in BRAF(V600E) melanomas.34 As noted above, suppression of NRAS expression in NRAS-mutant melanomas resulted in similar growth inhibitory effects.25 Thus, BRAF or NRAS oncogenic mutations appeared to elaborate a stringent melanoma tumor dependency through aberrant MAPK pathway activation.
In BRAF(V600E) melanomas, the mechanistic basis for oncogene dependency appears to involve both proliferative and apoptotic regulation. Oncogenic BRAF inhibits BIM, a pro-apoptotic member of the BCL-2 protein family,35 and suppression of BRAF activity facilitates translocation of the pro-apoptotic protein BMF to the cytosol in cells harboring the mutation.36 Moreover, two downstream effectors of RAF signaling—ERK and RSK—were shown to phosphorylate and inhibit the LKB1 tumor suppressor. This results in an inability of LKB1 to activate AMP kinase, which normally down-regulates cell growth under various stress conditions.37 Thus, activated B-RAF and downstream MAPK effectors appear to augment multiple melanoma cell growth and survival pathways.
The emergence of selective RAF inhibitors has provided an opportunity to evaluate the “drugability” of BRAF and NRAS oncogene mutations in vitro and in the clinical setting. Toward this end, small molecule RAF inhibitors such as PLX4032 potently suppressed the growth of BRAF(V600E) melanoma cells, whereas this inhibitor had little effect on cancer cells lacking this mutation.38,39 Selective RAF inhibitors also suppressed the formation of BRAF(V600E) tumors in murine xenograft models.40 These data suggest that stratification of melanomas according to BRAF(V600E) status might be enriched for patients most likely to benefit from small molecule RAF or MEK inhibitors. Recent clinical trial results with selective RAF inhibitors have borne out this prediction. In a phase 1 trial of PLX4032, the overall response rate of BRAF(V600E) melanomas was 90%, with nearly 80% experiencing a partial response by Response Evaluation Criteria in Solid Tumors (RECIST) criteria.41 NRAS-mutant melanomas did not respond to this agent. Similar results have also been observed with other investigational RAF inhibitors. These initial observations raise the possibility that RAF inhibition may ultimately gain a prominent role in the clinical management of BRAF(V600E) melanomas.
In light of the substantial evidence for “drugable” RAF dependency described above, it was somewhat surprising that selective RAF inhibition showed minimal efficacy against NRAS-mutant melanomas both in vitro and in early clinical studies. Initial insights into this puzzling discordance emerged from several reports that showed that exposure of RAS-activated cancer cells to selective RAF inhibitors paradoxically increased MAPK signaling, as measured by increased p-MEK and p-ERK levels.42–44 Biochemical studies revealed that inhibitor-bound BRAF polypeptides retained an ability to form homo- or heterodimers with other (drug-free) RAF molecules, and that this dimerization was associated with MEK phosphorylation in the setting of RAS activation. These provocative results may help explain why some RAF inhibitors fail to inhibit MAPK signaling in cells lacking BRAF(V600E) mutations (e.g., RAS mutant cells). They may also provide a basis for the unexpectedly high incidence (25% to 30%) of keratocanthomas and squamous cell carcinomas that arise in patients treated with selective RAF inhibition.41
MEK
Since the MEK1/2 serine-threonine kinases transmit the critical MAPK signal downstream of RAS and RAF, considerable interest has also emerged regarding these kinases in melanoma biology and therapeutics. In contrast to NRAS and BRAF, MEK1/2 mutations are rare in melanoma (indeed, in cancer in general), although they have occasionally been reported.45 Nonetheless, pharmacologic MEK inhibition presented another possible therapeutic inroad into BRAF- or NRAS-mutant melanomas. Robust preclinical evidence favoring this notion derived from a genetic and pharmacologic analysis showing that various MEK inhibitory compounds demonstrated markedly enhanced potency against BRAF(V600E) cancer cells compared to cell lines lacking oncogenic MAPK pathway mutations.46 In contrast to studies with selective RAF inhibitors, at least some NRAS-mutant melanoma cells were also sensitive to MEK inhibitors, suggesting that the utility of MEK inhibition might extend beyond the BRAF(V600E) context in some cases.46
Although initial clinical trial results of MEK inhibition in melanoma were disappointing compared to the RAF inhibitor trials described above, more recent studies have yielded responses in patients with BRAF(V600E) mutations following MEK inhibitor treatment.47 In addition, molecular analyses have provided an initial glimpse into how mechanisms of resistance might emerge to targeted MAPK pathway inhibition. In one case, a single relapsing focus was resected from a patient who was otherwise responsive to the MEK inhibitor AZD624448; the resistant tumor was found to harbor a somatic MEK1 mutation capable of conferring resistance to both MEK and BRAF inhibition in vitro. These intriguing translational studies have also triggered trials of combined RAF and MEK inhibition as a means to augment the magnitude or duration of clinical responses in BRAF(V600E) melanoma patients. A comprehensive elaboration of resistance to both RAF and MEK inhibition may provide new insights into possible combination therapeutic trials directed against this genetically defined tumor subtype.
THE PHOSPHATIDYLINOSITOL 3-KINASE PATHWAY
The PI3K pathway operates mainly through the downstream activation of the survival kinase AKT (Fig. 27.3), which activates the growth-promoting mammalian target of rapamycin (mTOR) pathway and inhibits proapoptotic effectors such as caspase 9 and BAD. PI3K was capable of replacing N-RAS to induce invasive melanomas in a skin xenograft model.49 However, unlike the MAPK pathway, genetic alterations specifically targeting components of the PI3K signaling cascade do not occur at high frequency in melanoma.50
Several lines of evidence support the cooperative effects of MAPK and PI3K dysregulation in melanoma genesis. For example, transformation of immortalized human melanocytes required the combination of activated BRAF and PI3K, but not BRAF alone, in an artificial skin graft model system.26 More recently, concomitant expression of oncogenic BRAF together with PTEN silencing in an inducible melanocyte-specific mouse model resulted in robust melanoma development with 100% penetrance.30,51 These melanomas arose following a short latency and exhibited a high propensity for metastatic dissemination. Together, these data suggest that dysregulated BRAF and PI3K signaling may underpin melanoma genesis in certain subsets of melanomas. Toward this end, a recent study suggested that N-RAS-mutant melanomas may show enhanced reliance on RalGEF signaling,52 which can be operant downstream of RAS in several contexts.
PTEN
Of the PI3K pathway mutations that do occur, losses of chromosome 10q encompassing PTEN tumor suppressor is the most frequent, the caveat being that additional tumor suppressor(s) may reside in this region (see below). PTEN normally down-regulates phosphorylated AKT via suppression of the second messenger PIP3 (Fig. 27.3). In various genetically engineered mice bearing solid tumors, PTEN loss can be analogous to p53 inactivation, in that one or the other can be gating for oncogenesis. In melanoma, somatic point mutations and homozygous deletions of PTEN are uncommon. Although allelic loss of PTEN is observed only in about 20% of melanoma, loss of expression of PTEN is reported to be in the range of 40% of melanoma tumors,53 suggesting other mechanisms of its inactivation. Functionally, ectopic expression of PTEN in PTEN-deficient melanoma cells can abolish phospho-AKT activity, induce apoptosis, and suppress growth, tumorigenicity, and metastasis.50 Correspondingly, heterozygous germline or homozygous somatic inactivation of Pten in the mouse strongly promoted tumor formation in multiple cell lineages, a phenotype that was accelerated by the additional deletion of Ink4a/Arf.54 Significantly, these double mutants developed melanoma at low penetrance, consistent with the hypothesis that PTEN inactivation in human melanoma may require additional genetic events such as the commonly associated BRAF(V600E) mutation. As noted above, this hypothesis was recently borne out in a mouse model of simultaneous PTEN loss and oncogenic BRAF induction in melanocytes.51 The relevance of PTEN inactivation in NRAS-mutant melanomas is less clear; however, studies in genetically engineered mouse models raise the possibility that RAS and PTEN inactivation may also interact to promote melanoma genesis and metastasis.55
Many melanomas harbor chromosome arm level hemizygous deletions involving the PTEN locus (10q24), although not all are specific to PTEN.4 Thus, other tumor suppressors may exist in this genomic region. The Myc antagonist MXI represents one possibility, as Myc is amplified or overexpressed in RAS-induced Trp53-deficient melanomas in the mouse.16 Other candidate tumor suppressor genes from chromosome 10q include CUL2 and KLF6; both genes are significantly down-regulated in melanomas harboring 10q deletions,4 although functional evidence that they exert tumor suppressor roles remains scant.
AKT
Elevated phospho-AKT (indicative of activation) has been adversely associated with patient survival56 and is common in BRAF(V600E) melanomas.57 Elevated phospho-AKT was particularly manifest in melanoma brain metastases compared to other metastatic sites.57 Copy number gains of the AKT3 locus and rare AKT3 point mutations58 have been detected in melanomas, suggesting that the AKT signaling point itself may be oncogenic.59 Interestingly, targeted depletion of AKT3 could trigger apoptosis,59 while AKT1 behaved as a tumor suppressor in melanoma cell lines,60 raising the possibility of partially nonredundant functions of AKT isoforms.
RECEPTOR TYROSINE KINASES
Receptor tyrosine kinsases (RTKs) are a diverse family of transmembrane kinases that have been implicated in many neoplasms. Several RTKs map to known regions of recurrent melanoma DNA copy number gain or amplification, with corresponding alterations in their expression levels.
c-KIT
The c-Kit gene plays an essential role in melanocyte development, as does its ligand, stem cell factor. Mutation of either results in pigmentation deficiencies, and injection of c-Kit blocking antibody in mice was used to identify the presence of melanocyte stem cells within hair follicles.61 Numerous immunohistochemical studies have linked progressive loss of c-KIT expression with the transition from benign to primary and metastatic melanomas.62–64 Thus, at first glance, KIT appeared to be inactivated during melanoma genesis and progression.
However, KIT is somatically activated by point mutations in a specific subset of melanomas that arise within mucosal, acral, or chronic sun-damaged surfaces of the body.13 High-resolution amplicon melting analyses followed by direct DNA sequencing revealed that three of these mutations harbored a L576P mutation with selective loss of the normal allele.65 L576P is a known gastrointestinal stromal tumor–associated mutation that maps to the 5’ juxtamembrane domain where most activating KIT mutations cluster.66 Importantly, these observations suggest the potential utility of c-KIT targeted kinase inhibitors for this subset of previously incurable melanoma patients. Initial case reports have provided preliminary support for this notion.67 Clinical trials using imatinib are currently under way to determine whether this discovery may represent a major opportunity for targeted therapy in melanoma as previously shown for gastrointestinal stromal tumors.
EPIDERMAL GROWTH FACTOR RECEPTOR FAMILY
The epidermal growth factor receptor (EGFR) activates both the MAPK and PI3K pathways in many cell types. In melanomas, copy number gain of chromosome 7 (where the EGFR gene resides) is linked to overexpression of EGFR, despite the lack of focal amplifications.68 EGFR activation increased the number of visceral metastases in severe combined immunodeficiency (SCID) mouse xenografts without affecting melanoma growth in vitro.69 In the inducible H-RAS mouse model,26 transcriptomic analysis revealed up-regulation of EGF family ligands including amphiregulin and epiregulin.70 Furthermore, expression of a dominant negative form of EGFR abolished the tumorigenicity of RAS-driven melanoma cells, indicating that the MAPK pathway is dependent on an uncompromised EGFR signal.71
A sequencing-based study of the tyrosine kinome in melanoma found that ERBB4 mutations may affect as many as 20% of melanomas.72 Unlike other well-known oncogene mutations, the ERBB4 mutations identified in this study were mostly nonrecurrent (e.g., the same amino acid or conserved region was rarely affected); however, ERBB4 mutations correlated with increased activation of and dependence on the corresponding protein for viability. If validated by resequencing of large independent sample cohorts, targeted therapeutics against ERBB4 and perhaps other members of the EGFR family may warrant investigation in melanoma.
c-MET
The c-MET gene product and its ligand hepatocyte growth factor/scatter factor (HGF/SF) are known to activate the MAPK pathway, but both have many additional functions. Overexpression of c-MET and HGF is correlated with melanoma progression. Copy gains involving the c-MET locus at 7q33-qter are associated with invasive and metastatic cancers in humans,73 and elevated MET/HGF expression is correlated with metastatic ability in murine melanoma explants.47 HGF/SF overexpression in a transgenic mouse model triggered spontaneous melanoma formation after a long latency (up to 2 years); however, time to tumor onset was greatly reduced by exposure to UVB or Ink4a/Arf deficiency.74 Thus, HGF/MET signaling may play an important role in melanoma genesis and progression. The development of MET inhibitors should allow formal testing of this hypothesis in melanoma patients.
THE MITF PATHWAY
MITF encodes a lineage transcription factor whose function is critical to the survival of normal melanocytes. The identification of MITF amplification in melanoma defined this transcription factor as a central modifier of melanoma.75 In so doing, this discovery identified a novel class of oncogenes termed lineage survival oncogenes.76,77 That is, a tumor may “hijack” extant lineage survival mechanisms in the presence of selective pressures to ensure its own propagation. The elucidation of MITF as an oncogene took a cross-tissue approach, wherein the NCI-60 cell line panel representing nine tumor types was subjected to both gene expression and high-density single-nucleotide polymorphism (SNP) array analysis.75 A recurrent gain of 3p13–14 significantly segregated melanoma from other tumor classes, with MITF as the only gene in the region showing maximal amplification and overexpression. MITF amplification was subsequently detected in 10% of primary cutaneous and 15% to 20% of metastatic melanomas by fluorescence in situ hybridization, correlating with decreased survival in Kaplan-Meier analyses of 5-year patient survival. Exogenous MITF, in combination with activated BRAF, showed transforming capabilities in immortalized primary human melanocytes. Additionally, inhibition of MITF in cell lines showing 3p13–14 amplification reduced growth and survival and conferred sensitivity to certain anticancer drugs. MITF gene disruption leads to coat color defects in mice and pigmentation defects in humans, due to diminished viability of melanocytes. This suggested that MITF was essential for the lineage survival of melanocytes, supporting the contention that it is also critical for the survival of melanomas.
The downstream elements of the MITF pathway include both pigment enzyme genes as well as genes involved in proliferation and survival. MITF intersects with a number of established melanoma pathways, including the transcriptional activation of INK4A, c-Met, and CDK2.17 Moreover, MITF is activated downstream of both c-Kit and MAPK signaling, via ERK-directed phosphorylation (Fig. 27.3).78 Additionally, activated BRAF is known to target MITF for proteolytic degradation, which may select for refractory cellular variants harboring amplified MITF. Recently, the ETS transcription factor ETV1 was found to positively regulate MITF expression in melanoma, and ETV1 may function as an amplified melanoma oncogene in its own right.79 Furthermore, the mouse Bcl2 proapoptotic gene was shown to be a transcriptional target of MITF.80 Collectively, these observations place MITF in a central role of melanoma signal integration.
THE WNT PATHWAY
WNT signaling has long been implicated in a variety of cancers. Its activation of downstream transcriptional events has been hypothesized to control lineage commitment and differentiation fates as well as self-renewal properties. The WNT pathway has also been linked to major developmental decisions in neural crest derivatives, with a differentiation bias toward the melanocytic lineage.81 Additionally, the WNT pathway appears to intersect with MITF functionality at several levels, including direct interactions between MITF and the WNT downstream transcriptional coactivator LEF-1.82 WNT signaling may augment MITF expression, and MITF may mediate at least a portion of WNT functionality in melanoma.83
CTNNB1 and WNT5A
Stabilizing CTTNB1 mutations that render β-catenin resistant to proteolytic degradation promote tumorigenesis. Such mutations are uncommon in patient samples, despite a high incidence in melanoma cell lines.84 Nevertheless, a significant fraction of clinical melanoma lesions exhibit immunohistochemical evidence of nuclear β-catenin localization,85 a hallmark of WNT pathway activation. Additional evidence for the importance of the WNT pathway emerged from gene expression studies that identified a β-catenin-independent Wnt5a signaling pathway correlated with higher grades of clinical melanoma samples.86 Engineered overexpression of Wnt5a enhanced the in vitro invasiveness of melanoma cells, a property suppressed by blockade of the Wnt5a receptor Frizzled-5. Mechanistically, Wnt5a was shown to enhance the action of the PKC pathway, which is believed to control cell adhesion and motility.
THE MC1R PATHWAY
Pigmentation exerts a major influence on skin tumor susceptibility, as it is well documented that fair skin is more sensitive to UV radiation and melanoma genesis. The mechanism underlying this observation is partially explained by the protective effects of melanin, which is produced by melanocytes and distributed to interfollicular keratinocytes. Genetically, the red hair color/pale skin (RHC) phenotype is linked to variant alleles of the melanocyte-specific melanocortin 1 receptor gene (MC1R), which is central to melanin synthesis.87 The ligand for the G-protein–coupled MC1R is the MSH peptide, which activates downstream signaling consisting of a cAMP-CREB/ATF1 cascade, culminating in the induced expression of MITF. Not all individuals carrying RHC alleles have identical melanin production, yet increased risk for melanoma genesis remains notable regardless,88 implying that melanin-independent mechanisms might impact the susceptibility of RHC carriers. One possible node is cAMP, the MC1R as second messenger, which may activate pathways incompletely understood at present, such as MAPK and PI3K.89
Recent data have implicated the MSH/MC1R pathway in the normal UV pigmentation (tanning) response in skin, a response that is linked to skin cancer (and melanoma) risk in humans. A “redhead” mouse model (frameshift mutation in MC1R) was used to demonstrate that the UV-tanning response is dependent on MC1R signaling, because keratinocytes respond to UV by strongly up-regulating expression of MSH. The “fairskin” phenotype was rescued by topical administration of a small molecule cAMP agonist.90 The resulting dark pigmentation in genetically redhead mice was protective against UV-induced skin carcinogenesis. Subsequent analyses revealed that the p53 tumor suppressor protein may function as a “UV sensor” in keratinocytes, translating UV damage into direct transcriptional stimulation of MSH expression.91
THE FAK PATHWAY
NEDD9 was recently demonstrated to enhance the metastatic efficiency of both mouse melanomas and human cell lines by activation of the FAK pathway, which enhances cell motility and adhesion.92 The discovery of these properties of NEDD9 was enabled by the evolutionary conservation of genetic pathways, which highlighted significant genetic changes when mouse tumors were compared to human melanomas. Array comparative genomic hybridization analysis of metastatic and nonmetastatic tumors derived from the Ink4a/Arf−/− inducible H-RAS mouse melanoma model26 pinpointed an 850 kb minimal common region (MCR) of amplification on chromosome 13. Only one gene in this MCR, Nedd9, showed a significant up-regulation in metastatic mouse melanomas but not in normal melanocytes or nonmetastatic melanomas. The syntenic human region, 6p24–25, similarly undergoes copy number gain in 36% of human metastatic but not nonmetastatic melanomas.92 The cross-species comparison allowed the delimitation of a focal region of interest and designation of NEDD9 as a candidate target of 6p gain in humans.
NEDD9 appears to modulate metastasis activity in vitro and in vivo. In cell-based assays, invasiveness of human melanoma cells was enhanced or inhibited by overexpression or knockdown of NEDD9, respectively. Similarly, knockdown of Nedd9 in metastatic melanoma cells with Nedd9 amplification drastically inhibited distal metastasis to various organs from subcutaneous primary tumor sites. Furthermore, inhibition of FAK itself abolished the invasive potential conferred by NEDD9 in Boyden chamber assays, implicating the entire pathway in metastasis.92 NEDD9 expression was found to be up-regulated in 50% of primary melanomas when compared to benign nevi, raising the possibility that NEDD9 might confer other biological activities during the early stages of melanoma genesis and that elevated NEDD9 expression might predict a risk of future metastasis.
CANCER STEM CELLS AND MELANOMA
Evidence supporting the biological and therapeutic relevance of a precursor tumor stem cell population has been cited in several tumor types; however, the role of cancer stem cells in melanoma remains controversial. Enthusiasm for a melanoma stem cell model was augmented by a study of ABCB5, an adenosine triphosphate–binding cassette transporter whose homologues have been implicated in stem cell multidrug resistance.93 Cancer cells expressing high ABCB5 were found to correlate significantly with melanocytic lineage and multidrug resistance in the NCI-60 cell line set.93,94 Mechanistically, ABCB5 was shown to mediate efflux of the anticancer drug doxorubicin and resistance to melanoma cell killing by this agent via modulation of membrane potential. Notably, the tissue distribution of ABCB5 protein partially colocalized with the putative stem cell marker CD133, suggesting that the drug resistance phenotype might be most closely associated with a long-lived, refractory stem cell population.
Evidence challenging the melanoma stem cell model emerged from xenotransplantation studies in immunocompromised mice. Whereas numerous studies in support of cancer stem cells utilized nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice to show that only approximately 1 of 10,000 tumor cells was capable of tumor initiation, a more recent study employed a more severely immunocompromised strain (NOD/SCID interleukin-2 receptor gamma chain null [Il2rg2/2]).95 In this setting, more than 1 of 4 melanoma cells were capable of tumor initiation; furthermore, neither ABCB5 nor any other stem cell marker expression could stratify the cells based on tumorigenic efficiency.95 These results suggest that the “classic” tumor stem cell hypothesis may not fully explain melanoma tumorigenesis; on the other hand, it remains unclear which (if any) xenotransplantation context accurately models the tumor stem cell phenotype. Additional studies are therefore needed to determine the extent to which stem cell features modify melanoma biology or chemotherapeutic response.
CANCER GENOMICS AND TRANSLATION
Advances in sequencing technology have made it possible to obtain the complete genome sequence of entire cancer genomes at ever diminishing costs. Together with developments in computational biology, these advances promise to usher in a new understanding of cancer genome alterations and the tumorigenic mechanisms that result. One of the first cancer genomes to be sequenced was that of a cell line (and its paired normal counterpart) derived from a patient with metastatic melanoma.96 This effort uncovered more than 33,000 somatic base substitutions, of which 187 were nonsynonymous coding mutations. As expected, most base mutations were C→T transitions indicative of ultraviolet exposure. However, the distribution of mutations across the genome was uneven, with a diminished prevalence in “gene footprint” areas.96 This distribution suggested the presence of transcription-coupled DNA repair in these regions. Thus, the complete sequence of this melanoma genome highlighted the mutational and selection dynamics that gave rise to this tumor.
Other studies have investigated the global transcriptional alterations characteristic of melanoma by deploying similar sequencing technologies to characterize cDNA from melanomas (termed RNA-seq). One recent RNA-seq study of ten melanomas (eight “short-term” cultures and two cell lines) identified multiple gene fusions, chimeric transcripts, splice isoforms, and base mutations not previously described.14 Whereas the gene fusions derived from structural genomic alterations occurred uniquely, several chimeric “read-through” transcripts (transcripts consisting of exons from two adjacent genes) were recurrently detected in different tumors. One such read-through event involved CDK2, a kinase involved in melanoma cell division that is also a transcriptional target of MITF.14,97
The high UV-associated base mutation rate in melanoma suggests that nearly 2% of all genes may harbor nonsynonymous coding mutations in a typical cutaneous melanoma, most of which are likely to be “passenger” events with little biological consequence to melanoma genesis or progression. Thus, cataloging all significant genomic alterations that might represent “driver” events (see below) will require not only sequencing hundreds of tumor specimens but also the principled application of increasingly sophisticated analytical methods for data deconvolution. Several large-scale U.S. (Cancer Genome Atlas) and international efforts (International Cancer Genomic Consortium)98 have taken on the ambitious goal of comprehensively characterizing the genomes of diverse human tumors including melanomas. Thus, it seems certain that the next decade will witness major breakthroughs in melanoma genome characterization that inform the biology and treatment of this malignancy.
Distinguishing “driver” events that may confer transforming activity or dictate prognosis or response to emerging targeted therapeutics will also require intensive functional validation efforts.99 An example of this was the recent discovery of GOLPH3 as an oncogene in melanoma.100 Intersection of chromosomal copy number variations (CNVs) in melanoma with those observed in other tumors followed by multilevel functional and clinicopathological validation studies led to the identification of the GOLPH3 gene as a first-in-class Golgi oncogene in a region of recurrent copy number gain.100 Furthermore, through a broad range of mechanistic and biological studies drawing from model systems as diverse as yeast, mouse, and human, authors showed that GOLPH3 exerts its oncogenic activity in part through regulation of TOR signaling, and that tumor cells with a high level of GOLPH3 exhibited increased sensitivity to mTOR inhibition.100 In the aggregate, this example highlights the importance of systematic functional validation and detailed biological studies to realize the potential of cancer genomics.
Toward this end, genome-scale functional genetic screens with RNAi have also begun to yield insights in melanoma. For example a systematic RNAi screen found that loss of the secreted protein IGFBP7 could bypass senescence associated with ectopic BRAF(V600E) expression.101 Moreover, many BRAF(V600E) melanomas showed loss of IGFBP7.101 In another RNAi study, loss of GAS1 was found to promote melanoma metastases in a murine model, suggesting that GAS1 may function as a metastasis suppressor.102 While the larger clinical importance of these observations remains to be established, these studies illustrate the power of systematic functional screens to offer new insights into melanoma biology that may ultimately have therapeutic implications.
LOOKING AHEAD
The identification of genetic alterations associated causally or otherwise prominently with melanoma initiation and progression presents the opportunity to exploit the genome for molecular biomarkers of melanoma pathogenesis and progression as well as for targets of therapeutic intervention. The initial success of both targeted therapeutics and immunotherapies that exploit this burgeoning molecular knowledge have paved the way for future rational interventions and in particular therapeutic combinations that may enable more durable clinical responses for many patients with advanced melanoma.
Genome-wide assays for DNA copy number alterations, RNA expression patterns, and increasingly protein activation (phosphorylation) states will continue to inform molecular mechanisms and contribute to melanoma pathogenesis. Within the next several years, many new melanoma cancer genes and effector pathways will likely be discovered through large-scale genome sequencing efforts. Evolving technologies will further uncover additional roles of gene dysregulation, such as epigenetic modification (e.g., methylation) or noncoding RNAs such as microRNAs. The application of sophisticated computational analyses should link these myriad observations so as to allow discovery of cellular networks and modules that exert crucial roles in melanoma genesis, survival, and progression. Finally, application of these types of analyses to relapsing tumor specimens should elaborate the spectrum of mechanisms by which therapeutic resistance emerges, thereby informing additional treatment options. Altogether, the intersection of scientific discovery and therapeutic innovation offers great potential to reduce death and suffering from this malignancy.
Selected References
The full list of references for this chapter appears in the online version.
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010;60(5):277.
2. Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005;436(7051):720.
4. Lin WM, Baker AC, Beroukhim R, et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res 2008;68(3):664.
5. Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Gen 1994;8(1):15.
6. Chin L, Pomerantz J, Polsky D, et al. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Devel 1997;11(21):2822.
13. Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353(20):2135.
14. Berger MF, Levin JZ, Vijayendran K, et al. Integrative analysis of the melanoma transcriptome. Genome Res 2010;20(4):413.
15. Chin L. The genetics of malignant melanoma: lessons from mouse and man. Nat Rev Cancer 2003;3(8):559.
16. Bardeesy N, Bastian BC, Hezel A, et al. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol 2001;21(6):2144.
17. Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev 2006;20(16):2149.
22. Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet 2007;39(3):347.
24. Ackermann J, Frutschi M, Kaloulis K, et al. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res 2005;65(10):4005.
26. Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature 1999;400(6743):468.
27. Kabbarah O, Chin L. Revealing the genomic heterogeneity of melanoma. Cancer Cell 2005;8(6):439.
30. Dhomen N, Reis-Filho JS, da Rocha Dias S, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 2009;15(4):294.
31. Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res 2003;63(17):5198.
32. Wellbrock C, Ogilvie L, Hedley D, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res 2004;64(7):2338.
33. Karasarides M, Chiloeches A, Hayward R, et al. B-RAF is a therapeutic target in melanoma. Oncogene 2004;23(37):6292.
34. Hoeflich KP, Gray DC, Eby MT, et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res 2006;66(2):999.
37. Zheng B, Jeong JH, Asara JM, et al. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell 2009;33(2):237.
40. Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A 2008;105(8):3041.
41. Flaherty K, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;36(9):809–815.
42. Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010;140(2):209.
43. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464(7287):427.
44. Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464(7287):431.
46. Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006;439(7074):358.
48. Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A 2009;106(48):20411.
49. Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet 2005;37(7):745.
51. Dankort D, Curley DP, Cartlidge RA, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009;41(5):544.
57. Davies MA, Stemke-Hale K, Lin E, et al. Integrated molecular and clinical analysis of AKT activation in metastatic melanoma. Clin Cancer Res 2009;15(24):7538.
61. Nishimura EK, Jordan SA, Oshima H, et al. Dominant role of the niche in melanocyte stem-cell fate determination. Nature 2002;416(6883):854.
67. Hodi FS, Friedlander P, Corless CL, et al. Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol 2008;26(12):2046.
70. Bardeesy N, Kim M, Xu J, et al. Role of epidermal growth factor receptor signaling in RAS-driven melanoma. Mol Cell Biol 2005;25(10):4176.
71. Sibilia M, Fleischmann A, Behrens A, et al. The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell 2000;102(2):211.
72. Prickett TD, Neena SA, Xiaomu Wei, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet 2009;41(10):1127.
73. Bastian BC, LeBoit PE, Hamm H, Brocker E-B, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res 1998;58(10):2170.
75. Garraway LA, Widlund HR, Rubin MA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005;436(7047):117.
76. Garraway LA, Sellers WR. From integrated genomics to tumor lineage dependency. Cancer Res 2006;66(5):2506.
77. Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer 2006;6(8):593.
79. Jané-Valbuena J, Widlund HR, Perner S, et al. An oncogenic role for ETV1 in melanoma. Cancer Res 2010;70(5):2075.
80. McGill GG, Horstmann M, Widlund HR, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 2002;109(6):707.
81. Dorsky RI, Moon RT, Raible DW. Control of neural crest cell fate by the Wnt signalling pathway. Nature 1998;396(6709):370.
86. Weeraratna AT, Jiang Y, Hostetter G, et al. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002;1(3):279.
90. D’Orazio JA, Nobuhisa T, Cui R, et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 2006;443(7109):340.
91. Cui R, Widlund HR, Feige E, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 2007;128(5):853.
93. Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature 2008;451(7176):345.
96. Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463(7278):191.
100. Scott KL, Kabbarah O, Liang MC, et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 2009;459(7250):1085.
101. Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008;132(3):363.
102. Gobeil S, Zhu X, Doillon CJ, Green MR. A genome-wide shRNA screen identifies GAS1 as a novel melanoma metastasis suppressor gene. Genes Dev 2008;22(21):2932.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


