Congenital Dyserythropoietic Anemias
Gary Kupfer
Linette Bosques
Bertil Glader
The congenital dyserythropoietic anemias (CDAs) are a heterogeneous group of inherited blood disorders characterized by anemia and morphologic abnormalities of erythroid precursors in the bone marrow, a consequence of dyserythropoiesis and ineffective erythropoiesis.1, 2 These disorders principally affect the erythroid lineage, whereas other nonerythroid hematopoietic lineages seem to be unaffected. On the basis of morphologic abnormalities of the bone marrow, CDAs were classified into three different types: type I, type II, and type III. However, additional variants requiring further characterization have also been identified.3 Approximately 600 cases of CDA have been reported worldwide, CDA II being the most common.3 CDA I and II are inherited in an autosomal recessive pattern, and CDA III exhibits autosomal dominant inheritance and a sporadic form.4 The disease-related genes for CDA I, II, and III have been localized by linkage analysis, and the diseasecausing genes have been identified for CDA I and II.
CDA patients display marked ineffective erythropoiesis and dyserythropoiesis.5, 6 Ineffective erythropoiesis may lead to intermittent jaundice, splenomegaly, hepatomegaly, and iron overload.2, 7 Iron overload is also a result of blood transfusion therapy, although even transfusion-independent patients may develop iron overload due to increased iron absorption.8 Dyserythropoiesis results in a variety of dysplastic features of erythroblasts in the bone marrow: internuclear chromatin bridges, karyorrhectic nuclei, binuclearity, multinuclearity, vacuolation of the cytoplasm, and duplication of the plasma membrane.9 Diagnosis of CDA relies on light and electron microscopy analyses to identify the characteristic morphologic abnormalities of the erythroblasts in the bone marrow.10 Mutation analysis is becoming available for some CDAs.
CDA should be suspected in any individual with a chronic anemia that is largely due to ineffective erythropoiesis, as well as in individuals with unexplained iron overload. The presence of ineffective erythropoiesis is characterized by a low absolute reticulocyte count for the degree of anemia despite erythroid hyperplasia in the marrow. Further suggestive laboratory data include a low serum haptoglobin level and increased serum lactic dehydrogenase (LDH) activity. Generally, the anemia is mildly or moderately macrocytic in CDA I, normocytic in CDA II, and mildly macrocytic in CDA III.
CONGENITAL DYSERYTHROPOIETIC ANEMIA TYPE I (CDA I)
CDA I is inherited in an autosomal recessive pattern and it represents the second most common form of CDA.6 It is most prevalent among Western Europeans, Middle Easterners (Israeli Bedouins, Lebanese, Kuwaitis, Saudi Arabians), Indians, and Japanese, with more than 150 reported cases.2
Clinical Features
Observations on CDA I have been enhanced by studies on a large cluster of cases in Israeli Bedouin families11, 12, 13 and on cases in the German CDA registry.14 The anemia varies from mild to severe, with the most severe cases presenting in infancy and the milder cases presenting in adolescence or later in life. Some degree of anemia is present in about two thirds of neonates, often requiring transfusion, but less than 10% of cases remain transfusion dependent later in life.14, 15 Occasionally, severe anemia in the fetus causes hydrops fetalis with pericardial and pleural effusions and edema.16, 17 Jaundice and splenomegaly are common findings at all ages. Gallstones may develop and require cholecystectomy. In addition, distal limb malformations, such as syndactyly, absence of phalanges and nails, an additional phalanx, and duplication or hypoplasia of the metatarsals, have been reported in several cases of CDA I.18 Other congenital abnormalities found include café au lait spots, short stature, flattened vertebral bodies, hypoplastic rib, Madelung deformity, and deafness.2 Deterioration of vision with retinal angioid streaks and macular degeneration has been described in two cases.19, 20
Laboratory Features
CDA I patients usually have a macrocytic anemia with MCV values up to 115; normocytic CDA I patients still display macrocytes and thus an increased RDW. Anisocytosis, poikilocytosis (including teardrop-shaped poikilocytes), and basophilic stippling are other features of the peripheral blood smear (Fig. 40.1). Increased hemoglobin A2 levels and unbalanced globin chain synthesis are found in some cases in the absence of β-thalassemia mutations.13 Bone marrow examination reveals marked erythroid hyperplasia with megaloblastic erythroblasts. One of the defining features of CDA I is the presence of thin internuclear chromatin bridges between nearly completely separated erythroblasts in 0.6% to 2.8% of cells6, 10 (Fig. 40.2). In only one well-documented case were such bridges not seen.19 A small proportion of erythroblasts are binucleate and occasional cells have three or four nuclei. Other abnormalities include RBC basophilic stippling, irregular nuclear outlines, and karyorrhexis.
A second defining structural feature of CDA I is multiple rounded electron-lucent areas within the electron-dense heterochromatin seen in up to 60% of erythroblasts, giving the nucleus a spongy or “Swiss cheese” appearance10, 21 (Fig. 40.3B,C). In some erythroblasts, there are also marked invaginations or evaginations of the nuclear envelope, and the invaginations carry cytoplasm and cytoplasmic organelles such as mitochondria into the nucleus (Fig. 40.3C).22 Bone marrow macrophages contain phagocytosed morphologically abnormal erythroblasts (Fig. 40.4).
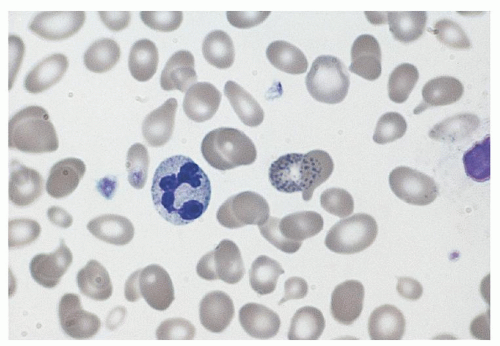 FIGURE 40.1. Blood smear from a patient with congenital dyserythropoietic anemia type I. (From Wickramasinghe SN. Congenital dyserythropoietic anemias. In: Wickramasinghe SN, McCullough J, eds. Blood and bone marrow pathology. Edinburgh: Churchill Livingstone, 2003:273-282, with permission.) |
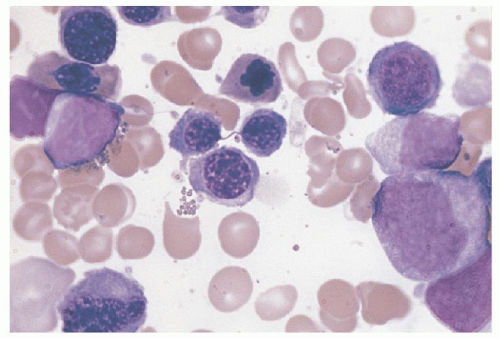 FIGURE 40.2. Bone marrow smear of a patient with congenital dyserythropoietic anemia type I showing an internuclear chromatin bridge. (From Wickramasinghe SN. Congenital dyserythropoietic anemias. In: Wickramasinghe SN, McCullough J, eds. Blood and bone marrow pathology. Edinburgh: Churchill Livingstone, 2003:273-282, with permission.) |
Management
CDA I often follows an indolent course, although patients may require red cell transfusions in infancy, with infections, and during pregnancy. Only some patients benefit from splenectomy and this is in contrast to CDA II.2, 11, 14 Erythropoietin administration has no effect on the anemia of CDA I.22 Following the fortuitous observation that treatment of hepatitis C in a CDA I patient with interferon-α2a led to a substantial hematologic improvement, several additional patients, including a severely affected infant, have been treated with interferon-α2a, interferon-α2b, or peg- interferon-α2b, and these patients also have also experienced hematologic improvement.16, 24, 25, 26, 27, 28, 29 Interferon-α should be considered in the management of CDA I with severe anemia. In one case, long-term therapy was reported to reduce iron overload.29 Careful monitoring for secondary hemochromatosis is important, at times requiring iron chelation therapy.
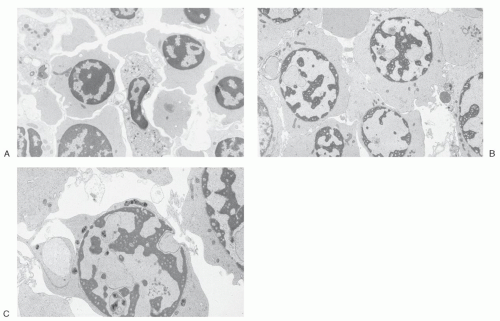 FIGURE 40.3. Ultrastructure of erythroblasts from a normal individual (A) and a patient with congenital dyserythropoietic anemia type i. B and C demonstrate the abnormal “Swiss cheese” appearance of the heterochromatin (compare with A), and C shows invagination of the nuclear membrane and nuclear membrane-bound areas of cytoplasm (containing iron-laden mitochondria) within the nucleus. (From Wickramasinghe SN. Congenital dyserythropoietic anaemias: clinical features, haematologic morphology and new biochemical data. Blood Rev 1998;12:178-200, with permission.) |
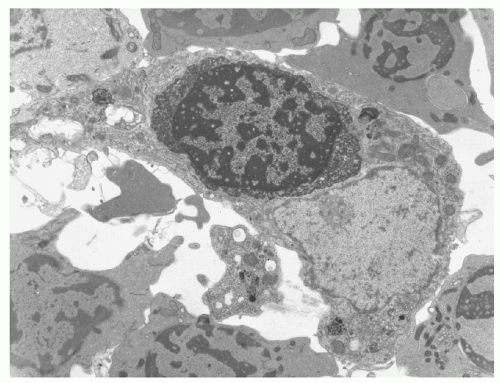 FIGURE 40.4. Bone marrow macrophage from a case of congenital dyserythropoietic anemia type I containing an ingested erythroblast with abnormal heterochromatin. |
Molecular Biology of CDA I
In 1998 the gene for CDA I was localized to chromosome 15q15.1-15.3.30 and it is now recognized that CDA I is caused by mutations in the CDAN1 gene.31 CDANI has 28 exons spanning 15 kb of genomic DNA and encodes a highly conserved protein of unknown function called codanin-1. Mutations in the CDAN1
gene are mainly located in the 3′ half of the gene and no homozygote for null type mutations has been identified, suggesting that the complete absence of codanin-1 may be lethal.32 One founder missense mutation is observed mainly in Israeli Bedouins with CDA I which converts arginine to tryptophan at codon 1,040 and generates an NcoI restriction site.31 This same missense mutation was found in 11 individuals with CDA I of two unrelated Lebanese families. A single mutation in CDAN1 has been found in several sporadic cases and in one case, no mutations in CDAN1 were found.31
gene are mainly located in the 3′ half of the gene and no homozygote for null type mutations has been identified, suggesting that the complete absence of codanin-1 may be lethal.32 One founder missense mutation is observed mainly in Israeli Bedouins with CDA I which converts arginine to tryptophan at codon 1,040 and generates an NcoI restriction site.31 This same missense mutation was found in 11 individuals with CDA I of two unrelated Lebanese families. A single mutation in CDAN1 has been found in several sporadic cases and in one case, no mutations in CDAN1 were found.31
Analysis of codanin-1 orthologs revealed the Drosophila homolog, dlt, is required for cell survival and cell cycle progression through the S phase.33 Interestingly, codanin-1 increases during the S phase and is phosphorylated and excluded from condensed chromosomes during mitosis.34 This study demonstrated that codanin-1 localizes to the nucleus, specifically to heterochromatin, during interphase and it is regulated by E2F1 transcription factors.34 Another study confirmed codanin-1 localization to the nucleus, but it revealed that it is more abundant in the cytoplasm.35 Codanin-1 mutations lead to the abnormal accumulation of heterochromatin protein HP1α in the Golgi apparatus of CDA I erythroblasts but not normal erythroblasts.35 However, overall chromatin structure as indicated by genomewide epigenetic marks of several histone modifications is normal in CDA I erythroblasts.
Recently, codanin-1 was shown to interact with histone chaperone Asf1, which regulates the import of histones during DNA replication.36 Codanin-1 knockdown increases the amount of chromatin-bound Asf1, enhancing DNA synthesis. Conversely, forced expression of codanin-1 interferes with Asf1 function due to Asf1 sequestration in the cytoplasm. Thus codanin-1 acts as a negative regulator of replication.
This close association with heterochromatin suggests that codanin-1 may be involved in mechanisms controlling gene transcription and chromatin structure remodeling. Thus, a clear understanding of the biochemical behavior of codanin-1 may provide us insight into how this interaction is established.
CONGENITAL DYSERYTHROPOIETIC ANEMIA TYPE II (CDA II)
Type II CDA was first categorized as such by Heimpel and Wendt in 1968. It is the most common type of CDA, and several hundred patients have been described.3 The mode of inheritance is autosomal recessive.
Clinical Features
CDA II is usually a normocytic anemia with evidence of ineffective erythropoiesis and premature peripheral red cell destruction, causing jaundice and hepatosplenomegaly. In very mild cases, the anemia may be so slight as to remain undiscovered until late in adult life, although a majority of patients have a hemoglobin less than 11 g/dl. Overall, a third of affected children require transfusion in the first year of life, but transfusion requirements decrease in subsequent years, and only 5% of patients need regular transfusions in adulthood.37 As in other congenital disorders in which ineffective erythropoiesis is a prominent feature, such as β thalassemia major, complications of extramedullary hematopoiesis may occur, such as dysmorphic facies, particularly in severely affected individuals. Cholelithiasis and secondary hemochromatosis are frequent complications, with iron overload the presenting manifestation in some cases.38 Occasionally, transient red cell aplasia secondary to parvovirus B19 infection may develop.39 Mental retardation,3, 40 hemihypertrophy,3 and piebaldism and vaginal atresia have all been reported.41
Related posts:
 Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Erythrocytosis
Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Erythrocytosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


