Dale N. Gerding, Vincent B. Young Keywords antibiotic-associated colitis; antibiotic side effects; C. difficile infection; clostridial toxin; Clostridium difficile; fecal microbial transplant; fecal transplant; geriatric infections; health care–associated infection; metronidazole; microbiome; NAAT; nosocomial infection; oral vancomycin; pseudomembranous colitis; recurrent C. difficile infections; spores
Clostridium difficile Infection
The administration of antibiotics can be complicated by a number of unintended consequences, of which gastrointestinal side effects are quite common. Gastrointestinal symptoms occur in up to 25% to 50% of patients, depending on the specific antimicrobial agent, patient population, and epidemiology.1 Although the majority of these side effects are mild, consisting of minor antibiotic-associated diarrhea without systemic signs of illness, some patients can develop frank colitis and severe clinical manifestations, including toxic megacolon, intestinal perforation, sepsis, and death. Clostridium difficile infection (CDI) is believed to be responsible for about 25% of all cases of antibiotic-associated diarrhea and is the underlying cause in nearly all cases of severe disease.
Although Koch’s postulates were fulfilled for C. difficile nearly 40 years ago,2 we still have an incomplete understanding of how disruption of the normal indigenous gut microbiota leads to C. difficile colonization and frank disease. Additionally, whereas adequate antimicrobial therapy for CDI was described shortly after the etiologic role for the pathogen was established,3 a recent increase in the apparent severity, prevalence, and recurrence of CDI4,5 has prompted additional interest in understanding the pathogenesis of this infection.
Historical Overview
Prior to 1978 the disease pseudomembranous colitis (PMC) and the organism C. difficile had never been linked. PMC was described in humans by Finney in 18936 and in animals in 1943 by Hambre and colleagues after administration of penicillin that resulted in rapid animal death.7 PMC occurred in patients prior to the antibiotic era, perhaps confirming why no prior antibiotic use is found in a small subset of current CDI patients.8 However, it was the antibiotic era that was accompanied by an increase in PMC, particularly in association with use of lincomycins.9,10 It was the publication by Tedesco and co-workers10 in 1974 that started the search for a cause for PMC linked to clindamycin use and at the time was called clindamycin-associated colitis. Of 200 clindamycin-treated patients, 21% were found to have diarrhea and nearly half of those (10%) were found to have PMC, including 38% of patients who inexplicably had onset of diarrhea after clindamycin had been stopped.
After 4 years of intensive research, in 1977 and 1978 a succession of papers leading to the identification of C. difficile and its toxins as the cause of clindamycin-associated colitis or PMC and of vancomycin as an effective treatment were published. Laboratories of Larson in the United Kingdom and of Bartlett, Fekety, and Finegold in the United States were largely responsible for these discoveries, using the hamster as an essential animal model for CDI.11–17
In retrospect, the rapidity and thoroughness of the CDI research discoveries of the 1970s may have engendered an early perception that there was little else to learn. This may have led to a clinical perception of CDI as a nuisance disease that was not even monitored as a health care–associated infection in many hospitals. This perception of CDI changed markedly in the early 21st century when CDI epidemics in North America and Europe with high mortality occurred that were caused by a previously little known C. difficile strain that has been variously described as North American pulsed-field type 1 (NAP1), restriction endonuclease analysis (REA) group BI, and polymerase chain reaction (PCR) ribotype 027, collectively referred to as NAP1/BI/027.5,18
Prior to the realization that C. difficile was the etiologic agent for the majority of cases of PMC other infectious agents had been implicated as causative agents of PMC and antibiotic-associated diarrhea.19 Staphylococcus aureus was one of the first bacteria associated with PMC after the administration of antibiotics.20,21 Experimental evidence was provided by studies that demonstrated that antibiotic-treated chinchillas would develop enterocolitis after infection with S. aureus.22 Although C. difficile is responsible for most cases of severe antibiotic-associated colitis, enterotoxigenic S. aureus strains can still be isolated from C. difficile–negative cases.23 Enterotoxigenic Clostridium perfringens strains have also been isolated from patients with antibiotic-associated diarrhea.24 Another form of severe antibiotic-associated colitis (associated with hemorrhage but not pseudomembrane formation) appears to be due to infection with Klebsiella oxytoca.25 These findings indicate that a proportion of cases of antibiotic-associated colitis and diarrhea that are due to infection are not secondary to C. difficile, but this is only in the minority of cases. Additionally, it needs to be restated that infection (including infection with C. difficile) accounts only for about a fourth of cases of antibiotic-associated diarrhea. In noninfectious cases of antibiotic-associated diarrhea, disruption of the normal composition and function of the indigenous gut microbiota alone, as detailed in the following section, is believed to be the underlying cause.26
Pathogenesis
The Indigenous Gut Microbiota and Antibiotics
The pathogenesis of antibiotic-associated diarrhea and colitis, whether or not it is associated with CDI, is believed to center on the disruption of the normal, indigenous gut microbiota by antimicrobial administration. The concept of the indigenous gut microbiota playing a central role in human health has existed for at least 100 years. Recent scientific and technologic advances have permitted the study of the role that the gut microbiome plays in human health and disease,27,28,29 including the development of antibiotic-associated diarrhea and colitis.
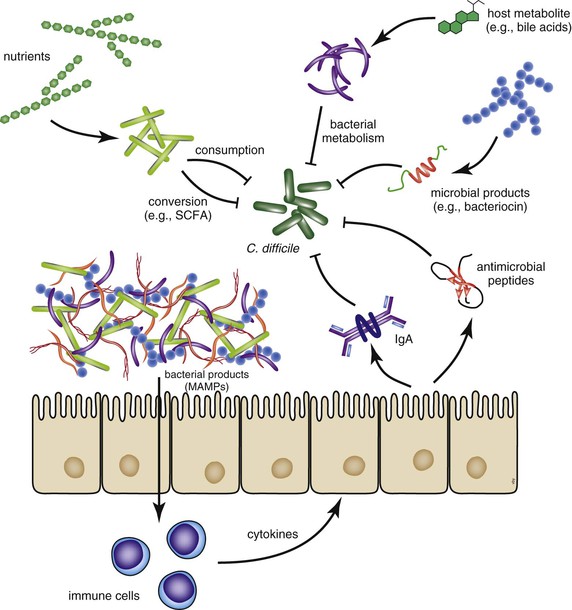
The indigenous gut microbiota are believed to play a number of critical roles in host homeostasis; and for the purposes of CDI, the function of importance has been called “colonization resistance.”30 Colonization resistance refers to the ability of the normal gut microbial community to resist the ingrowth of pathogenic microbes. A number of different mechanisms have been speculated to be important for maintaining colonization resistance, including competition for nutrients, occupancy of ecologic and physical niches, production of antimicrobial products, and signaling through the host immune system (Fig. 245-1).31 Regardless of the specific mechanisms, therapeutic antibiotic administration can profoundly disrupt the indigenous gut microbiota community and therefore disrupt colonization resistance. Studies in human and animal systems indicate that antibiotics can have long-lasting effects on the gut microbiome structure and thus alter gastrointestinal function.32–35 With regard to C. difficile the altered gut microbiome structure and function can directly influence the biology of the pathogen in terms of growth and pathogenesis. In the following sections, as specific topics in the pathogenesis and virulence of C. difficile are discussed, potential influences of the indigenous microbiota will be highlighted.
Sporulation and Germination in Clostridium difficile
As with many anaerobic bacteria, during periods of environmental stress vegetative C. difficile bacteria initiate the sporulation program, which results in the production of the spore form of the organism.36,37 This physically robust form is stable to oxygen stress, temperature extremes, and desiccation. Within the hospital environment, this includes resistance to the effects of alcohol-based hand sanitizers.38 The spore form thus represents a reservoir for the organism, which can persist in the environment.
After ingestion of spores, germination is apparently triggered by the environment that exists within the gastrointestinal tract.39 It has been shown that primary bile acids, including taurocholic acid, can serve as potent germinants in vitro, with glycine functioning as a co-germinant.40–42 Interestingly, members of the indigenous microbiota efficiently metabolize primary bile acids through the process of deconjugation and dehydroxylation to produce secondary bile acids,43–45 some of which have been shown to be inhibitory to C. difficile.41,42 Therefore, one mechanism by which the disruption of the indigenous microbiome by antibiotics can lead to CDI is through alteration of in vivo bile acid metabolism.31 This leads to a situation that favors germination of ingested spores and the subsequent vegetative outgrowth of the pathogen. The key role of spore germination in the pathogenesis of C. difficile may serve as an attractive therapeutic target, as has been recently demonstrated with the use of a synthetic bile salt analogue that inhibits germination and has a protective effect in animal studies.46
Toxin Production
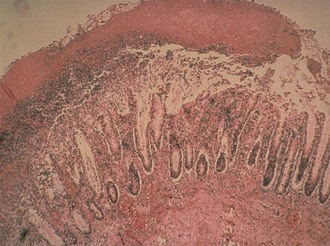
The histopathologic hallmark of CDI is damage to the mucosal epithelium with generation of an acute, neutrophil-predominant inflammatory response with the formation of a pseudomembrane consisting of sloughed epithelial cells, inflammatory cells, and a fibrinous exudate (Fig. 245-2). Damage to the epithelium is caused by the most well-characterized C. difficile virulence factor, the large glucosyltransferase toxins TcdA and TcdB.47–49 Members of the large clostridial toxin family, TcdA and TcdB are produced within the gastrointestinal lumen during the stationary phase of vegetative growth of C. difficile. These toxins are taken up by the cells of the mucosal epithelium through the process of receptor-mediated endocytosis.50,51 Within the endosome the toxin is autocatalytically cleaved to release the active A subunit, which is transferred to the host cell cytoplasm.52 Subsequently, the active subunit glycosylates host cell guanosine triphosphatases belonging to the Rho family of cytoskeletal regulatory proteins.53,54 Intracellular activation of Rho guanosine triphosphatases leads to disruption of the actin cytoskeleton, ultimately leading to apoptotic and necrotic cell death. In cultured epithelial cells, cytoskeletal disruption results in cell rounding and cytotoxicity. In vivo these effects are manifested by loss of cell-cell tight junctions with subsequent increase in epithelial permeability.55,56 TcdA and TcdB also induce the secretion of cytokines in host cells, including interleukin-8, which likely leads to the acute neutrophilic inflammatory infiltrate that characterizes CDI.57
Other Virulence Factors
Some strains of C. difficile secrete a toxin that is a member of the clostridial binary toxins exemplified by the iota toxin from Clostridium perfringens.58 C. difficile binary toxin (CDT) is a two-part toxin that is an adenosine diphosphate ribosyltransferase that is specific for actin monomers, thus disrupting the actin cytoskeleton.58 CDT is produced by only a small fraction of C. difficile strains, and thus its role in pathogenesis is unclear.59 Although some reports suggest that infection with a CDT-producing C. difficile strain results in more severe clinical disease, this is not a universal finding and the actual significance of infection with CDT-producing strains is not clearly defined.60–62
In addition to the three toxins, multiple other potential virulence factors for C. difficile have been studied, including surface layer proteins, surface polysaccharides, flagella, and various adhesins.63 To date, the specific roles of any of these factors in C. difficile virulence have not been delineated, but ongoing work on these factors may lead to new strategies to prevent and treat CDI.
Host Response to Clostridium difficile Infection
Patients infected with C. difficile can mount adaptive immune responses to the toxins TcdA and TcdB.64 The level of antibody response to the C. difficile toxins is inversely correlated with the relative risk of developing recurrent disease.65 Additionally, because the pathogenesis of C. difficile is tightly linked to pathogen expression of TcdA and TcdB, immunotherapy either in the form of toxin-based vaccines or passive administration of toxin-specific antibodies is being developed (see later).
Additional work has been done examining the role of innate immune responses in the pathogenesis of CDI.66 In animal models, mice that are deficient in Toll-like receptor (TLR) signaling, including MyD88 knockout mice, were shown to be more susceptible to experimental CDI.67,68 Similarly, animals deficient in signaling through the intracellular pattern recognition pathway involving Nod1 are more susceptible to CDI.69 It appears that signaling through the innate immune system early during infection is important in host defense against C. difficile. For example, stimulation through the TLR5 pathway that senses bacterial flagellin can improve host survival in experimental CDI.70
Pathogenesis of Recurrent Clostridium difficile Infection
Whereas the majority of patients with symptomatic CDI respond to antibiotic therapy directed against the pathogen, up to 25% of patients will have recurrent symptoms after the discontinuation of C. difficile therapy for an initial episode of disease.71,72 The risk for recurrence increases with each episode, resulting in significant morbidity in this population of patients. A number of factors have been investigated with regard to the pathogenesis of recurrent infection. As noted previously, the adaptive host response may play a role in determining the risk for recurrent disease.65
Several studies have indicated that pathogen characteristics may contribute to the risk for recurrence. Infection with the epidemic NAP1/BI/027 C. difficile strain is associated with an increased risk for recurrence.73 The reasons for this are unclear, but they may have to do with the intrinsic antibiotic resistance (e.g., quinolone resistance), the dynamics of spore biology of the strain, or the presence of an additional toxin (i.e., binary toxin).
Because alteration of the indigenous gut microbiota by antibiotics is a prerequisite for the majority of cases of CDI, the role of the gut microbiome in recurrence has been examined. It has been shown that patients with recurrent CDI exhibit decreased microbial diversity of their indigenous gut microbes.74 Presumably, if the gut microbiota is unable to return to its baseline state, the patient is at increased risk for reinfection or regrowth of residual C. difficile after therapy. This has led to the exploration of restoring microbiome diversity through the administration of probiotic bacteria or the transplantation of feces from normal donors (see “Therapy”).
Altered Virulence in Specific Clostridium difficile Lineages
The recent increase in the incidence and apparent severity of CDI over the past 15 years has been associated with the widespread appearance of the NAP1/BI/027 strain of C. difficile. This has prompted a number of investigators to determine if this particular strain of C. difficile is actually associated with worse outcomes in infected patients and whether this strain has specific virulence determinants not widespread in other strains of the pathogen.
There are several factors that are characteristic of NAP1/BI/027 strains. These strains typically are resistant to newer fluoroquinolone antibiotics.5 Furthermore they possess the binary toxin CDT and also harbor a characteristic mutation in the anti-sigma factor tcdC, which is theorized to play a role in the regulation of toxins TcdA and TcdB.5 This mutation in tcdC introduces a frameshift that results in a truncated, nonfunctional protein.75 Because studies have demonstrated that these epidemic strains can produce increased levels of toxin in vitro, it has been proposed that this characteristic mutation is responsible for this phenotype, although this has been questioned by reconstitution of tcdC that showed no effect on toxin production.77 There is in vitro evidence that the TcdC regulator can inhibit in vitro expression of the large C. difficile toxins,75 but the presence of alterations within this regulatory gene does not directly correlate with increased production of toxins within clinical isolates.76 One group demonstrated that complementation of the tcdC mutation in trans will decrease the amount of toxin produced by a NAP1/BI/027 clinical isolate and reduce the virulence of this strain in the hamster model of infection.78 However a second group that utilized a genetic system that allowed correction of the tcdC mutation directly on the chromosome failed to show an association between tcdC genotype and the level of toxin production.77
Additional in vitro studies suggested that NAP1/BI/027 strains had an increased ability to sporulate,79 which could contribute to the ease with which these strains can spread within the health care environment; however, this observation has also been disputed by measuring sporulation in vitro in multiple C. difficile strains, including multiple NAP1/BI/027 strains that did not reveal increased sporulation of these epidemic strains.80 Perhaps most important to the enhanced virulence of these strains is the presence of a third toxin, binary toxin CDT, an adenosine diphosphate–ribosylating toxin that has been shown to enhance the lethality of toxin A in the hamster model and to cause a modest degree of hamster mortality in the absence of toxins A and B.81 This constellation of characteristics has also arisen within an unrelated epidemic strain of PCR ribotype 078 that was identified in Europe82 and together with ribotype 027 was found to exhibit increased 14-day mortality in patients with CDI.83
Despite this evidence, there is still debate over the relative importance of these genetic and phenotypic characteristics of the epidemic C. difficile strains in the pathogenesis of the organism.84 In spite of these conflicting data it is likely that our understanding of the basic pathogenesis of CDI will result in the development of more effective strategies for prevention and treatment of this infection.
Epidemiology
C. difficile is a ubiquitous organism, able to survive for long periods in the environment through sporulation. al Saif and Brazier showed the widespread presence of C. difficile in environmental sites, such as surface water, drinking water, swimming pools, and soil, and in a wide variety of animals, including dogs, cats, horses, sheep, and pigs.85 In addition, foods have been shown to be contaminated with C. difficile spores that are of the same ribotype that cause clinical disease. This includes beef, pork, turkey, and a variety of vegetables. It is highly likely that humans ingest C. difficile spores frequently but remain asymptomatic (and uncolonized) as a result of the “colonization resistance” of an intact gut microbiota.
Health Care–Associated Clostridium difficile Infection
It has been known since 1979 that the hospital environment is contaminated with C. difficile spores.86 In addition, C. difficile contamination of the hands of hospital personnel and of the home environment of patients and spore persistence for up to 20 weeks in the environment have been known since 1981.87 This environmental contamination is presumed to be the result of the repeated CDI diarrheal episodes of hospitalized patients and resistance of spores to killing by environmental cleaners and disinfectants other than bleach. In 1986, the first prospective case-control study of CDI was published and showed 87% of 149 cases to be health care associated (and hospital associated).88 In addition, C. difficile was found asymptomatically in the stool of 21% of control patients. Both the specific use of clindamycin and use of multiple antibiotics for the treatment of infection were identified as risk factors. McFarland and associates,89 in a landmark paper, used rectal swab specimens to identify hospital acquisition of C. difficile by 21% of sampled patients, 63% of whom were asymptomatic. Johnson and colleagues90 also used weekly rectal swab cultures to document health care (in this case, hospital) acquisition of C. difficile by 21% of patients. In their article they reported that 82% of patients were asymptomatic. The use of REA typing identified 18 unique REA types of C. difficile, but symptomatic disease (CDI) was caused only by two closely related REA types, B1 and B2, which also caused asymptomatic colonization in some patients but CDI in others.
It has also been shown that admission of asymptomatic patients carrying C. difficile in their stool preceded the acquisition of that specific strain of C. difficile by other patients on the same hospital ward in 85% of acquisitions.91 Although asymptomatic patients colonized with C. difficile may be a source of transmission to other patients, contrary to intuitive thinking, they have a significantly lower risk for developing CDI themselves compared with uncolonized patients on the same hospital wards at the same time.92 It is presumed that these asymptomatic colonized patients are protected from CDI either because they harbor nontoxigenic strains of C. difficile (which lack toxin genes and do not cause symptoms) or they are protected from the effect of C. difficile toxins through adaptive immunity (most likely an antibody response) if they are colonized by toxigenic strains of C. difficile.
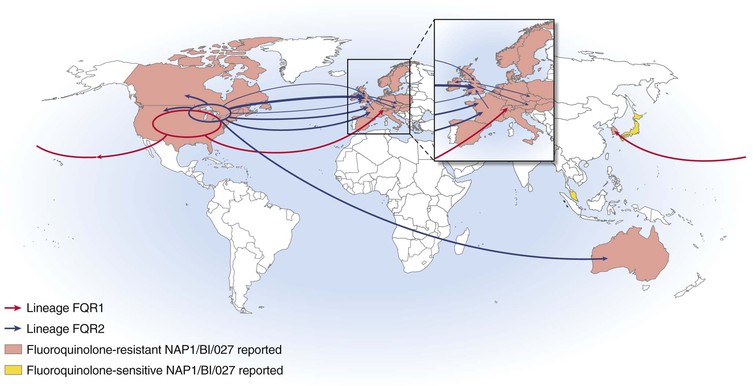
The health care epidemiology of CDI changed markedly in the early years of this century when multiple hospitals, first in the United States and then in Canada, reported widespread outbreaks of CDI of particularly high severity, causing high mortality and requiring increased use of colectomy to treat patients refractory to medical management.5,18,93 These outbreaks were caused by a specific group of C. difficile strains designated NAP1/BI/027 that rapidly spread to the United Kingdom and European Union countries. Although unknown at the time, there were two distinct lineages of NAP1/BI/027, designated FQR1 and FQR2, that acquired fluoroquinolone resistance mutations and a highly related conjugative transposon.94 The FQR1 lineage almost certainly originated in the United States and was widely disseminated in the United States with spread to Asia and Switzerland (Fig. 245-3). The FQR2 lineage was found in both Montreal in Canada as well as multiple U.S. sites and spread more widely than FQR1 to the United Kingdom and Europe. Health care–associated CDI incidence rose to as high as 22.5 CDI cases per 1000 discharges in the Montreal NAP1/BI/027 outbreak, with 30-day attributable mortality of 6.9%.18 More recent reports suggest that the current hospital incidence in the United States is in the range of 5 to 10 CDI cases per 10,000 days of care.95
Community-Associated Clostridium difficile Infection
There has never been a question that CDI occurs in the community setting, but whether the incidence of community-associated CDI versus community-onset CDI is increasing is an active epidemiologic question that is likely being affected by shorter inpatient stays and increased health care delivery in the outpatient setting. In particular, the use of antibiotics in the outpatient setting, recently calculated to amount to 258 million courses in 2010,96 is likely to create enormous pressure for the development of community-associated CDI. Hirschhorn and colleagues97 found the rate of community-acquired CDI to be 7.7 cases/yr/100,000 population in a health plan database in Boston published in 1994. No antibiotic exposure was found in a surprising 35% of patients in the previous 42 days. Similar rates of community-associated CDI (7.6 and 6.9 community-associated CDI cases per 100,000 population) were found in two surveys by the Centers for Disease Control and Prevention (CDC) in 2005 to 2006 in Philadelphia and Connecticut, respectively.98 Conflicting community rates have been found in other large databases. One of these, the U.K. General Practice Research Database, found that CDI increased from less than 1 case/100,000 in 1994 to 22 cases/100,000 in 2004. Only 39% of these patients had prior antibiotic exposure documented within 90 days before CDI.99 A reanalysis of the same data using a definition of CDI as treatment with oral vancomycin found an association with prior antibiotic exposure in 55% of patients.100 Using a more definitive methodology, a U.K. study of positive stool cultures from community-onset diarrhea patients showed 2.1% of stools positive for C. difficile toxin by cell cytotoxin assay in urban and rural patient cohorts, corresponding to rates of 29.5 and 20.2 cases/100,000 population, respectively, much higher than previously documented rates.101 Only 52% of these patients had antibiotic exposure in the previous 4 weeks.
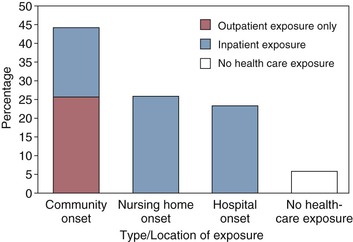
A more recent report of community CDI epidemiology conducted by the CDC through its Emerging Infections Program has clarified a number of aspects of CDI in the community and health care settings.95 In contrast to the 1986 report of 87% hospital-onset CDI, the more recent population-based report from CDC of 10,342 CDI cases in 111 acute-care hospitals and 310 nursing homes revealed only 25% hospital-onset CDI, a similar proportion with nursing home onset, and 45% with community-onset CDI (Fig. 245-4). Among all CDI cases, 94% had either inpatient or outpatient health care exposure within the previous 12 weeks, leaving only 6% of all CDI patients with no prior health care exposure.
In a related study, of 989 community-associated CDI patients, only 18% had no outpatient health care exposures and 41% had low-level and 41% high-level outpatient exposures.102 Of these, 358 (36%) did not receive antibiotics, but the incidence of antibiotic exposure was significantly higher in the groups with health care exposure than in those without (P < .01), an indication that health care exposure predisposes to CDI via exposure to antibiotics. The frequency of exposure to proton-pump inhibitors was not significantly higher in patients without antibiotic exposure than in patients with antibiotic exposure, suggesting that proton-pump inhibitor use is not an independent or surrogate (for antibiotic use) risk factor for community-acquired CDI. CDI patients with low-level health care exposures were more likely to be exposed to infants aged younger than 1 year (odds ratio [OR], 2.1; 95% confidence interval [CI], 1.1 to 4.5) and household members with CDI (OR, 6.9; 95% CI, 5.6 to 56.5) compared with patients with high-level exposures, suggesting that these are C. difficile exposure sources. There was no association between food or animal exposure and level of health care exposure, leaving the source for acquisition of C. difficile undetermined for most patients.
Molecular Epidemiology of Clostridium difficile Infection and Importance of Clostridium difficile Strain Type
The lack of a universally sensitive and reproducible typing system has slowed progress in the molecular epidemiology of CDI, but PCR ribotyping, pulsed-field gel electrophoresis (PFGE), and REA are the most frequent typing systems in use. REA was used to document the surprising finding that approximately 50% of isolates from second episodes of CDI were different from the original isolate, suggesting an environmental source for recurrent CDI at least half of the time,103 an observation that has shifted recently toward more frequent identification of the same strain as the cause of recurrence in over 80% of instances.104 REA was also used to document the wide diversity of C. difficile strains in one hospital (55 different types) during an endemic period.105 The REA J group found in multiple U.S. hospitals in the 1990s was shown by PCR ribotyping in the United Kingdom to be ribotype 001.106,107 These J/001 strains were clindamycin resistant, and increased CDI incidence was associated with clindamycin exposure in the patients. Molecular typing was instrumental in identifying the current epidemic C. difficile toxinotype III strains by both PFGE (type NAP1) and by REA (group BI) in hospitals in the United States and Canada and confirmed in the United Kingdom and Europe as PCR ribotype 027.5,18 These strains are highly resistant to fluoroquinolone antibiotics and are associated with fluoroquinolone use as a risk factor.18
Clostridium difficile Infection in Children
Hall and O’Toole first described C. difficile in 1935 as a colonizing organism in neonates.108 Ironically, this continues to be a contentious diagnostic issue in children up to the age of 1 to 2 years because the frequent asymptomatic colonization of these young children confounds the diagnosis of diarrhea because if tested they frequently will be positive for toxigenic strains of C. difficile; however, the frequency of stool positivity for C. difficile in these children with diarrhea is no higher than that of children of the same age without diarrhea.109 It is not known how very young children can tolerate colonization by toxigenic C. difficile without developing diarrhea or other symptoms. Data from neonatal rabbits suggests that there may be insufficient toxin A receptors in these young animals that allows them to tolerate toxin, but similar observations in humans have not been made.110 When children are sampled on a monthly basis over the first year of life, all are found to at some time be colonized with C. difficile, both toxigenic and nontoxigenic strains, and they are noted to frequently change strains over time.111 Because of the high rate of colonization in children younger than the age of 1 year, it is not recommended that they be tested for CDI.110 Beyond the age of 3 years the colonization rate of children resembles that of adults (<3%) and risk factors for CDI are also similar to those in adults, but CDI rates are generally much lower in children than in adults, especially adults older than age 65 years. Health care–associated CDI in children is associated with prolonged hospitalization and increased mortality, whereas community-associated CDI is not.112
Risk Factors
Risk factors have been identified for CDI, for recurrent CDI, for CDI severity, and for CDI caused by the epidemic NAP1/BI/027 strain.
Clostridium difficile Infection
The overall risk for developing CDI is dominated by antimicrobial exposure, including duration, number, and class of antimicrobial agents (Table 245-1).113 Risk is highest during therapy and in the first month after cessation of antimicrobial therapy and decreases between 1 and 3 months after therapy.114 In addition, stomach acid–inhibiting agents such as proton-pump inhibitors and histamine type 2 antagonists have also been associated with increased risk for CDI, although some studies fail to make this association.115 It is likely that the combination of an antimicrobial agent and a proton-pump inhibitor increase the risk for CDI.116,117 Patient factors are also important CDI risks, with advanced age, immunosuppression, prior hospitalization, and severity of underlying illness contributing to increased risk.118 A meta-analysis and strain risk comparisons have identified fluoroquinolone exposure and age older than 65 years as risks for CDI caused by NAP1/BI/027.119,120
TABLE 245-1
Risks for Development of Clostridium difficile Infection

Recurrent Clostridium difficile Infection
Risk factors for recurrent CDI are similar to those for an initial episode but are particularly predictable because one antecedent episode of CDI increases risk with each subsequent recurrence (Table 245-2). Additional antimicrobial treatment either during or after the initial CDI episode markedly increases risk for recurrence; and, as with primary CDI, the most elderly patients and those with the most severe underlying disease are at highest risk for recurrence.121,122,123
TABLE 245-2
Risk Factors for Development of Recurrent Clostridium difficile Infection

Severe Clostridium difficile Infection
There has been an ongoing search for a clinical prediction rule or score that would predict at the time of diagnosis which patients are destined for a severe CDI outcome, usually defined as death, need for intensive care unit admission, megacolon, perforation, shock, or requiring colectomy for treatment. A number of patient predictors such as a white blood cell (WBC) count greater than 15,000 or 20,000/µL and a rise in creatinine concentration of greater than 1.5 times baseline (or a serum creatinine level ≥1.5 mg/dL) have correlated with more severe disease.4,124,125 Low serum albumin level and elevated WBC count have been found to predict CDI severity independent of infecting strain type, but higher 14-day mortality has been observed with infection caused by two strains of C. difficile—NAP7-8/BK/078 and NAP1/BI/027—as well as with presence of specific patient biomarkers: elevated WBC count and C-reactive protein and low serum albumin concentration.84,126 Additionally, infection with the NAP1/BI/027 strain of C. difficile has resulted in lower cure rates and higher recurrence rates compared with other C. difficile strains when treated with either fidaxomicin or vancomycin.127 Although several studies have demonstrated the association between infection with NAP1/BI/027 strains and increased disease severity, this has not been a universal finding.84
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


