HEREDITARY SYNDROMES ASSOCIATED WITH CHILDHOOD CANCER PREDISPOSITION
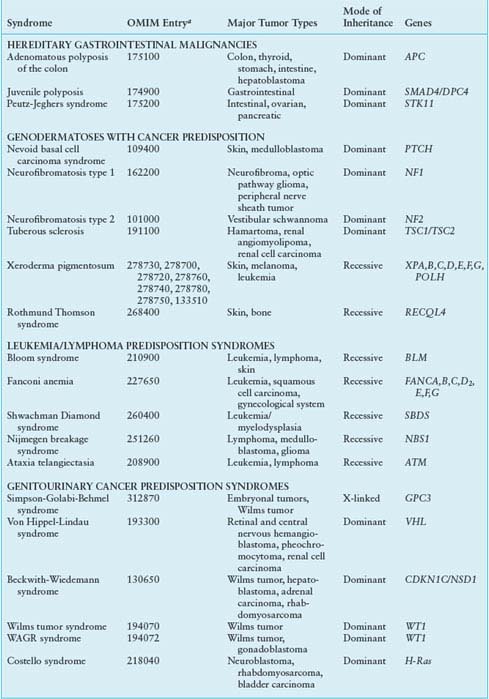
Whereas acquired alterations of dominant oncogenes most commonly occur in somatic cells, mutant tumor suppressor genes may be found either in germ cells or somatic cells. In the former, they may arise de novo or be transmitted from generation to generation within a family. The diversity of functions, cellular locations, and tissue-specific expression of the tumor suppressor genes suggest the existence of a complex, yet coordinated, cellular pathway that limits cell growth by linking nuclear processes with the intra- and extracytoplasmic environment. This discussion is limited to those genes for which pediatric tumors are frequently associated.
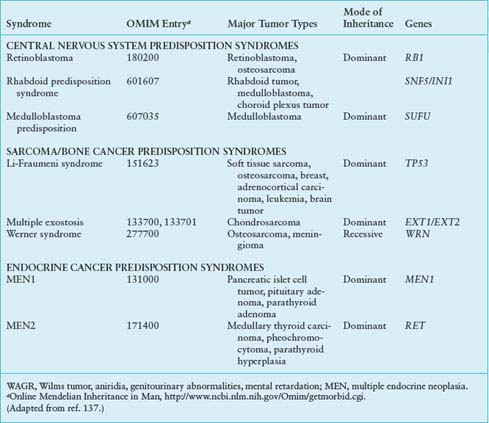
TABLE 29.2
COMMON CYTOGENETIC REARRANGEMENTS IN SOLID TUMORS OF CHILDHOOD
RETINOBLASTOMA: THE PARADIGM
Retinoblastoma is the prototype cancer caused by mutations of a tumor suppressor gene. It is a malignant tumor of the retina that occurs in infants and young children, with an incidence of approximately 1:20,000.5 Approximately 40% of retinoblastoma cases are of the heritable form in which the child inherits one mutant allele at the retinoblastoma susceptibility locus (Rb1) through the germ line, and a somatic mutation in a single retinal cell causes loss of function of the remaining normal allele, leading to tumor formation. Tumors are often bilateral and multifocal. The disease is inherited as an autosomal dominant trait, with a penetrance approaching 100%.6 The remaining 60% of retinoblastoma cases are sporadic (nonheritable), in which both Rb1 alleles in a single retinal cell are inactivated by somatic mutations. As one can imagine, such an event is rare, and these patients usually have only one tumor that presents itself later than in infants with the heritable form. Fifteen percent of unilateral retinoblastoma is heritable6 but by chance develops in only one eye. Survivors of heritable retinoblastoma have a several 100–fold increased risk of developing mesenchymal tumors such as osteogenic sarcoma, fibrosarcomas, and melanomas later in life.7 It is thought that several genetic mechanisms may be involved in elimination of the second wild type Rb1 allele in an evolving tumor. These mechanisms include chromosomal duplication or nondisjunction, mitotic recombination, or gene conversion.8
The Rb1 gene was eventually mapped to chromosome 13q14.9 Using Southern blot analysis, it was then possible to demonstrate that the second target gene that led to disease was actually the second copy of the Rb1 locus. Reduction to homozygosity of the mutant allele (or loss of heterozygosity [LOH] of the wild type allele) would lead to the loss of functional Rb1 and account for tumor development.
Using classic cloning techniques, a 4.7-kb complementary DNA fragment was isolated from retinal cells.10 This gene, Rb1, consisted of 27 exons and encoded a 105-kD nuclear phosphoprotein. As well as being altered in retinoblastoma, this gene and its protein product have been found to be altered in osteosarcomas, small cell lung carcinomas, and bladder, breast, and prostate carcinomas.10,11 Rb1 plays a central role in the control of cell-cycle regulation, particularly in determining transition from G1 through S (DNA synthesis) phase in virtually all cell types.
Although it is clear that Rb1 and its protein product play some role in growth regulation, the precise nature of this role remains obscure. In the developing retina, inactivation of the Rb1 gene is necessary and sufficient for tumor formation.12 It is now clear, however, that these tumors develop as a result of a more complex interplay of aberrant expression of other cell-cycle control genes. In particular, a tumor surveillance pathway mediated by Arf, MDM2, MDMX, and p53 (see later discussion) is activated after loss of Rb1 during development of the retina. Rb1-deficient retinoblasts undergo p53-mediated apoptosis and exit the cell cycle. Subsequently, amplification of the MDMX gene and increased expression of MDMX protein are strongly selected for during tumor progression as a mechanism to suppress the p53 response in Rb1-deficient retinal cells.13 Not only do these observations provide a provocative biological mechanism for tumor formation in retinoblastoma, but it also offers potential molecular targets for novel therapeutic approaches to this tumor.14,15 Although the Rb1 gene is expressed in virtually all mammalian tissues, only in the retina is its inactivation sufficient for tumor initiation. On the other hand, some Rb1 mutations appear to lead to an attenuated form of the disease, an observation that highlights the variable penetrance in families.16,17 Outside the retina, Rb1 inactivation is often a rate-limiting step in tumorigenesis generated by multiple genetic events. The molecular characteristics and potential functional activities of Rb1 are outlined in detail elsewhere in this volume.
The patterns of inheritance and presentation of retinoblastoma have been well described and the responsible gene identified. The basic mechanisms by which the gene is inactivated are understood, and provocative evidence indicates that the intricate functional interactions of pRB with its binding partners and other cell cycle targets will provide targets for development of novel small molecule therapies.
WILMS TUMOR: THREE DISTINCT LOCI
Wilms tumor, or nephroblastoma, is an embryonal malignancy that arises from remnants of an immature kidney. It affects approximately 1:10,000 children, usually before the age of 6 years (median age at diagnosis, 3.5 years). Five percent to 10% of children present with either synchronous or metachronous bilateral tumors. A peculiar feature of Wilms tumor is its association with nephrogenic rests, foci of primitive but nonmalignant cells whose persistence suggests a defect in kidney development. These precursor lesions are found within the normal kidney tissue of 30% to 40% of children with Wilms tumor. Nephrogenic rests may persist, regress spontaneously, or grow into a large mass that simulates a true Wilms tumor and presents a difficult diagnostic challenge.18 Another interesting feature of this neoplasm is its association with specific congenital abnormalities, including genitourinary anomalies, sporadic aniridia, mental retardation, and hemihypertrophy. The WT1 tumor suppressor gene is reduced to homozygosity, at least in part, in a small but highly informative set of sporadic Wilms tumors. In addition, sporadic and hereditary Wilms tumors have been described in which WT1 is specifically altered.
A genetic predisposition to Wilms tumor is observed in two distinct disease syndromes with urogenital system malformations: the WAGR (Wilms tumor, aniridia, genitourinary abnormalities, mental retardation) syndrome and the Denys-Drash syndrome (DDS)19 as well as in BWS, a hereditary overgrowth syndrome characterized by visceromegaly, macroglossia, and hyperinsulinemic hypoglycemia.20 These congenital disorders have now been linked to abnormalities at specific genetic loci implicated in Wilms tumorigenesis.
The WAGR syndrome has been correlated with constitutional deletions of chromosome 11q13.21 Whereas it is now known that the WAGR deletion encompasses a number of contiguous genes, including the aniridia gene Pax6,22 the cytogenetic observation in patients with WAGR was also important in the cloning of the WT1 gene at chromosome 11p13.23–25 Characterization of WT1 demonstrated that this gene spans approximately 50 kb of DNA and contains ten exons. The WT1 protein is a transcription factor. However, the identity of the gene(s) targeted by WT1 during normal kidney development is not known.
The second syndrome closely associated with this locus was initially described by Denys in 1967 and recognized as a syndrome by Drash 3 years later.26,27 DDS is a rare association of Wilms tumor, intersex disorders, and progressive renal failure.27 It has been demonstrated that virtually all patients with DDS carry WT1 point mutations in the germ line.28
WT1 is altered in only 10% of Wilms tumors. This observation implies the existence of alternative loci in the etiology of this childhood renal malignancy. One such locus also resides on the short arm of chromosome 11, telomeric of WT1, at 11p15. This gene, designated WT2, is associated with BWS. Patients with BWS are at increased risk of developing Wilms tumor, as well as other embryonic malignancies, including rhabdomyosarcoma (RMS), neuroblastoma, and hepatoblastoma.29 The putative BWS gene maps to chromosome 11p15 and is tightly linked to the Ha-ras oncogene homologue HRAS-I and the insulinlike growth factor-2 gene (IGF-2). Whether the BWS gene and WT2 are one and the same or two distinct yet closely linked genes remains to be determined. Other genes, including CDKN1C (p57KIP2), a maternally expressed gene that encodes a cyclin-dependent kinase inhibitor and negatively regulates cell proliferation,30 show aberrant methylation in tumors that are associated with cell-cycle deregulation. However, CDKN1C is rarely mutated in Wilms tumors.31 Thus, the search for other genes linked to Wilms tumor continues. Using long-oligonucleotide array comparative genomic hybridization (array CGH), a novel gene termed WTX was identified on chromosome Xq11.1. WTX is inactivated in one-third of Wilms tumors, and tumors with WTX mutations lack WT1 mutations. Whereas autosomal tumor suppressor genes undergo biallelic inactivation, WTX is inactivated by a monoallelic “single-hit” event that targets the single X chromosome in Wilms tumors in males and the active X chromosome in tumors from females.32 This observation suggests a more important role of the X chromosome in human cancers than had previously been appreciated.33
Although linkage studies have indicated that the gene for familial Wilms tumor must be distinct from WT1 and WT2, and from the gene that predisposes to BWS, to date, this gene has been neither cytogenetically localized nor isolated. Whether, of course, the gene for familial Wilms tumor interacts with the gene product of either of the two Wilms tumor suppressor genes has yet to be determined.
Finally, loss of the long arm of chromosome 16 has been observed in approximately 20% of Wilms tumor samples.34 This observation implicates yet another genetic locus in Wilms tumor. Linkage studies have generally also excluded this locus as the “familial” Wilms tumor gene.35 However, it is plausible that alterations at 16q can initiate tumorigenesis or be implicated in subsequent steps in the progression of malignancy.
Although both tumors represent classic models of the Knudson “two-hit hypothesis” of tumor development, the spectrum of genetic alterations in Wilms tumor is quite different from that in retinoblastoma. In the latter, there is strong evidence that a single gene is involved whose function is mediated through related cell-cycle control pathway genes, whereas a series of genetic alterations, or at least distinct genetic events, is required for Wilms tumorigenesis. Second, the “single-hit” mechanism of WTX inactivation suggests a novel basis for Wilms tumorigenesis; and third, unlike retinoblastoma, which is not associated with other developmental anomalies or congenital abnormalities, patients with Wilms tumor commonly exhibit a spectrum of nonmalignant urogenital, skeletal, and cardiac congenital abnormalities that suggest a dual role of the genes involved in both tumor formation and normal embryonic development.
NEUROFIBROMATOSES
The neurofibromatoses (NFs) comprise two similar entities. NF1 is one of the most common autosomal dominantly inherited disorders, affecting approximately 1 in 3,500 people,36 and half of them arise from new spontaneous mutations. Carriers of mutant NF1 are predisposed to a variety of tumors, including optic nerve glioma, neurofibroma and neurofibrosarcoma, malignant schwannoma, or malignant peripheral nerve sheath tumor, astrocytoma, and pheochromocytoma.37 Occurring with less frequency are leukemias, osteosarcoma, RMS, and Wilms tumor and pediatric gastrointestinal stromal tumors.38
Using standard linkage analysis, the NF1 gene was mapped to chromosomal band 17q11.2 and subsequently cloned.39 The NF1 gene is composed of 60 exons spanning 350 kb of the genome and is unusual in that it contains three embedded genes—OMGP, EV12A, and EV12B—of unknown function.40 The NF1 gene encodes a 2,818-amino acid protein, termed neurofibromin, which is ubiquitously expressed. One region of the gene shows extensive structural homology to the guanosine triphosphatase–activating domain of mammalian guanosine triphosphatase–activating proteins: loss of the protein’s activity results in failure of hydrolysis of guanosine triphosphate to guanosine diphosphate by the ras oncoprotein. Loss of neurofibromin function usually results from mutations in one allele of the gene, leading to premature truncation of the protein, followed by absence or mutations of the second allele in tumors. More than one mechanism appears to exist whereby malignant tumors develop in patients with NF.
In addition to structural alterations of both alleles of the NF1 gene, alternative splicing leading to dysregulation at the level of transcription has also been demonstrated. It appears that the two types of resulting protein may modify the modulation of RAS-regulated signal transduction. This loss of function is thought to lead to elevated levels of the guanosine triphosphate–bound RAS protein that transduces signals for cell division. NF1 is now considered to be one of the RASopathies, a class of developmental disorders caused by activation of the RAS-MAPK pathway.41 Loss of neurofibromin also is associated with activation of mammalian target of rapamycin (mTOR), suggesting a potential therapeutic target for NF1-associated tumors.42 NF type 2 (NF2) is much less frequent than NF1, occurring in only one in one million persons. Although it is also inherited as an autosomal dominant disorder with high penetrance, the new mutation rate in NF2 is low.43 It is clinically characterized by bilateral acoustic neuromas, spinal nerve root tumors, and meningiomas.
The NF2 locus was mapped to chromosome 22, band q12,44 and its 69-kD encoded protein, termed merlin, has been shown to be expressed in various tissues, including brain, although not as ubiquitously as NF1.45 The mechanism of tumor formation in NF2 appears to be in concordance with the Knudson two-hit model, although the mechanism of action of the NF2 protein has not yet been elucidated. Merlin is a member of the ERM (ezrin-radixin-moesin) family of proteins that links cell surface proteins to the cytoskeleton.46 Although ezrin expression has been linked to metastatic behavior,47 merlin appears to compete with ezrin activation,43 and merlin deficiency seems to enhance metastases and promote tumorigenesis through destabilization of adherens junctions.48 Vestibular schwannomas, a hallmark of NF2, have been shown to signal through epidermal growth factor receptor family receptors, suggesting a potential target for medical therapy in these difficult to manage tumors.49
NEUROBLASTOMA
Nonrandom chromosomal abnormalities are observed in more than 75% of neuroblastomas,50 and many of these are also found in neuroblastoma-derived cell lines. The most common of these is deletion or rearrangement of the short arm of chromosome 1, although loss, gain, and rearrangements of chromosomes 10, 11, 14, 17, and 19 have also been reported. The allelic losses indicate loss of function of as yet unknown tumor suppressor genes in these regions. It is believed that a tumor suppressor gene that lies on band p36 of chromosome 1 is critically important in the pathogenesis and aggressive nature of neuroblastoma. It has been shown that the loss of chromosome 1p is a strong prognostic factor in patients with neuroblastoma, independent of age and stage.51 Although it is as yet unclear which gene(s) in this region may be directly implicated in neuroblastoma development, aberrant expression of one candidate—p73—which is a member of the p53 tumor suppressor family, has been suggested to play a role in the neuroblastoma cell growth as well as chemotherapy resistance.52 p73 gives rise to multiple functionally distinct protein isoforms as a result of alternative promoter utilization and alternative mRNA splicing.53,54 Alternative splicing of the p73 mRNA results in more than seven protein isoforms that differ in the coding sequences of the COOH terminus (TA-p73 α, β, γ, δ, ε, ζ, η).
In addition to these COOH-terminal splice forms, three additional forms, Np73α, DNp73β, and DNp73β, are transcribed from an alternative promoter located in intron 3. Their protein products lack the NH2-terminal transactivation domain and are thus called DNp73. The full-length forms that contain the NH2-terminal transactivation domain are denoted TA. Higher levels of DNp73 are associated with an overall worse clinical prognosis, presumably because of the “antiapoptotic” properties of DNp73 and its ability to inactivate both TAp73 and p53.55,56
Two other unique cytogenetic rearrangements are highly characteristic of neuroblastoma.57 These structures, homogeneous staining regions and double-minute chromosomes, contain regions of gene amplification. The N-myc gene, an oncogene with considerable homology to the cellular protooncogene c-myc, is amplified within homogeneous staining regions and double-minute chromosomes. Virtually all neuroblastoma tumor cell lines demonstrate amplified and highly expressed N-myc,58 and N-myc amplification is thought to be associated with rapid tumor progression. Expression of N-myc is increased in undifferentiated tumor cells compared with much lower (or single-copy) levels in more differentiated cells (ganglioneuroblastoma and ganglioneuroma). N-myc expression is diminished in association with the in vitro differentiation of neuroblastoma cell lines.59 This observation formed the basis for current therapeutic trials demonstrating a survival advantage to patients treated with cis-retinoic acid.60 Furthermore, a close correlation exists between N-myc amplification and advanced clinical stage.61
Although it is clear that altered expression of N-myc contributes to the development of malignancy, it is not yet apparent which cellular functions are altered. The molecular mechanisms underlying regulation of neuroblastoma differentiation may be explained in part through the contribution of other genes and proteins. This is currently under intense investigation through the use of gene expression profiling of N-myc–positive versus –negative tumors.
Neuroblastoma cells that express the high-affinity nerve growth factor receptor trkA62 can be terminally differentiated by nerve growth factor and may demonstrate morphologic changes typical of ganglionic differentiation. Tumors showing ganglionic differentiation and trk gene activation have a favorable prognosis.62 In contrast, trkB receptor expression is associated with poor-prognosis tumors and appears to mediate resistance to chemotherapy.63,64 Resistance to multidrug chemotherapeutic regimens (multidrug resistance) is characteristic of aggressive, poorly responsive N-myc–amplified neuroblastomas. It is interesting to note that expression of the multidrug resistance–associated protein, found to confer multidrug resistance in vitro, is increased in neuroblastomas with N-myc amplification and decreased after differentiation of tumor cells in vitro.65 It has been demonstrated that high levels of multidrug resistance–associated protein expression are significantly associated with poor outcome, independent of N-myc amplification.65 Gain of chromosome segment 17q21-qter has been shown to be the most powerful prognostic factor yet.66 However, no gene has yet been implicated at this site.
A small subset of neuroblastomas is inherited in an autosomal dominant fashion. Until recently, the only gene definitively associated with neuroblastoma risk was PHOX2B, also linked to central apnea.67 De novo or inherited missense mutations in the tyrosine kinase domain of the ALK (anaplastic lymphoma kinase) gene on chromosome 2p23 have been observed in the majority of hereditary neuroblastoma families, as well as in somatic tumor cells.68–71 Current phase 1/2 clinical trials with ALK inhibitors substantiate the value of such target identification for novel therapies. The role of other molecular alterations in neuroblastoma continues to be elucidated. In addition to chromosomal loss on chromosome 1p36, unbalanced LOH at 11q23 is independently associated with decreased event-free survival. Alterations at 11q23 occur in almost one-third of neuroblastomas, being most commonly associated with stage 4 disease and age at diagnosis greater than 2.5 years. Both 1p36 LOH and 11q23 LOH were independently associated with decreased progression-free survival in patients with low- and intermediate-risk disease.72
Yet another valuable biological marker of clinical significance is telomerase expression and telomere length. In particular, short telomere length is predictive of favorable prognosis, irrespective of disease stage, while long or unchanged telomeres are predictive of poor outcome.73,74 Telomerase expression, as measured by telomerase reverse transcriptase (hTERT), has been shown to be negative in good-risk neuroblastoma, although it is high in tumors with unfavorable histology.74 The combined use of these markers—chromosomes 1p and 11q, N-Myc amplification, trkA and telomerase expression—as prognostic indicators provides a powerful armamentarium with which to develop rational stratified treatment programs for neuroblastoma.
EWING SARCOMA FAMILY OF TUMORS
Ewing sarcoma (ES) is one of the first examples in which the application of molecular diagnostics led to improved tumor classification. ES was first described by James Ewing75 as a bone tumor characterized by small, blue, round cells and minimal mitotic activity. Turc-Carel et al.76 identified a recurring reciprocal t(11;22) chromosomal translocation in these tumors in 1983. Investigators subsequently demonstrated a cytogenetically identical t(11;22) in adult neuroblastoma or peripheral primitive neuroectodermal tumor (pPNET), so named because of its histologic similarity to neuroblastoma.77 Based on the presence of the identical translocation, it was hypothesized that pPNET was related to ES. This translocation breakpoint has been molecularly characterized as an in-frame fusion between a new ES gene, EWS, on chromosome 22 and an ETS transcription family member, FLI-1, on chromosome 22.78–80
In addition to this fusion transcript being identified in pPNET, other variants, notably the chest-wall Askin tumor and soft tissue ES—previously treated as an RMS because of its location in soft tissue—were also shown to bear the identical fusion transcript. In total, five translocations also have been identified, invariably fusing the EWS gene to an ETS family member.81–84 More than 90% of the ES family of tumors (ESFTs) carry the EWS-ETS fusion gene, and a search for EWS-ETS by either reverse transcriptase-polymerase chain reaction or fluorescence in situ hybridization should be considered standard practice in the diagnostic evaluation of suspected ESFTs. Interestingly, although it was suggested that the specific fusion protein expressed in ESFT has prognostic significance,85 several prospective studies in the United States and Europe demonstrated no prognostic impact.86,87 The nature of the novel fusion transcription factor and its downstream targets is currently under intense investigation. One target of the EWS-ETS fusion is repression of the transforming growth factor-β type II receptor,88 a putative tumor suppressor gene.
Expression profiling analysis has also revealed that p53 is transcriptionally up-regulated by the EWS-ETS fusion gene.89 This is of particular interest because it is now known that expression of EWS-ETS can lead to apoptosis, and that additional alterations such as loss of p53 or p16 signaling, or both, appear to be necessary components of EWS-ETS–induced transformation.90 Investigators have now taken advantage of RNA interference technology to inhibit EWS-FLI-1 in Ewing cell lines to identify genes regulated by the fusion in the proper context. Using this approach, NKX2.2 and NR0B1 have been found to be a target gene of EWS-FLI-1 that is necessary for oncogenic transformation.91,92 Recent findings suggest that GGAA microsatellites might mark genes that are up-regulated by EWS-FLI-1 binding.93
RHABDOMYOSARCOMA
The two major histologic subtypes of RMS, embryonal and alveolar, have unique histologic appearances as well as distinctive molecular genetic abnormalities, while sharing a common myogenic lineage. Embryonal tumors comprise two-thirds of all RMS and are histologically characterized by a stroma-rich spindle cell appearance. Alveolar tumors comprise approximately one-third of RMS and are histologically characterized by densely packed, small, round cells, often lining a septation reminiscent of a pulmonary alveolus, giving rise to its name. Both histologic subtypes express muscle-specific proteins, including α-actin, myosin, desmin, and MyoD,94,95 and they virtually always express high levels of IGF-2.96
At the molecular level, embryonal tumors are characterized by LOH at the 11p15 locus, which is of particular interest because this region harbors the IGF-2 gene.97 The LOH at 11p15 occurs by loss of maternal and duplication of paternal chromosomal material.98 Although LOH is normally associated with loss of tumor suppressor gene activity, in this instance LOH with paternal duplication may result in activation of IGF-2. This occurs because IGF-2 is now known to be normally imprinted; that is, this gene is normally transcriptionally silent at the maternal allele, with only the paternal allele being transcriptionally active.99 Thus, LOH with paternal duplication potentially leads to a twofold gene-dosage effect of the IGF-2 locus. Furthermore, in alveolar tumors in which LOH does not occur, the normally imprinted maternal allele has been shown to be re-expressed.100 Thus, LOH and loss of imprinting in this case may lead to the same functional result—namely, biallelic expression of the normally monoallelically expressed IGF-2. However, loss of an as yet unidentified tumor suppressor activity due to LOH also remains a possibility. HRAS oncogene is also located at 11p15, and germ line mutations of HRAS occur in Costello syndrome, another tumor within the RASopathy family. Patients with Costello syndrome have an increased incidence of embryonal RMS.101 These data suggest the possibility of cooperativity between HRAS mutations and UPD at 11p15 in the development of ERMS.102
Alveolar RMS is characterized by a t(2;13) (q35;q14) chromosomal translocation.103 Molecular cloning of this translocation has identified the generation of a fusion transcription factor, fusing the 5’ DNA-binding region of PAX-3 on chromosome 2 to the 3’ transactivation domain region of FKHR gene on chromosome 13.104 A variant t(1;13) (q36;q14) has been identified in a smaller number of alveolar RMS tumors that fuse the 5’ DNA-binding region of the PAX-7 gene on chromosome 1 with the identical 3’ transactivation domain of the FKHR (Foxo 1A) gene.105 Fluorescence in situ hybridization or reverse transcriptase-polymerase chain reaction can be used to identify these PAX-FKHR fusions. In a review of 171 patients entered into the Intergroup Rhabdomyosarcoma Study Group IV study, the gene fusion was found only in alveolar RMS cases. In the 78 alveolar RMS cases, 55% were PAX3-FKHR+, 22% were PAX7-FKHR+, and 23% were fusion-negative.106 It has now become clear that these fusion-negative alveolar RMS tumors are clinically and molecularly indistinguishable from embryonal RMS, thus making the presence of the PAX-FKHR fusion a diagnostic criteria for alveolar RMS.107 The nature of this fusion-derived novel transcription factor and its downstream targets is the subject of active investigation. It also has been suggested that, like ESFT, in which the specific expressed fusion transcript has prognostic significance, the PAX-3–FKHR and the PAX-7–FKHR fusions lead to distinct clinicopathologic entities.108 The critical role of PAX-3-FKHR in the generation of alveolar RMS has now been recapitulated in a mouse PAX-3-FKHR knock-in model, coupled with either p53 or CDKN2a inactivation.109
The PAX-3–FKHR fusion is associated with increased expression of c-met.110 Met is the receptor tyrosine kinase for hepatocyte growth factor/scatter factor and is overexpressed in embryonal and alveolar RMS.111 A mouse model of embryonal RMS has been generated by expressing a hepatocyte growth factor transgene in Ink4a/Arf–/– mice. The tumors appear to arise from hyperplastic satellite cells (myoblastic precursor cells).112 The putative role of satellite cells in the pathogenesis of embryonal RMS is supported by a report demonstrating high PAX-7 expression in embryonal RMS compared to alveolar RMS and the association of PAX-7 expression with satellite cells.113 Other frequently reported genetic alterations that may be common to embryonal and alveolar RMS include activated forms of N- and K-RAS,114 inactivating p53 mutations,115 and amplification and overexpression of MDM2, CDK-4, N-MYC,116 and FGFR4 activating mutations.117
HEREDITARY SYNDROMES ASSOCIATED WITH TUMORS OF CHILDHOOD
Li-Fraumeni Syndrome
A few hereditary cancer syndromes are associated with the occurrence of childhood as well as adult-onset neoplasms. The paradigm LFS cancer was first described in 1969 from an epidemiologic evaluation of more than 600 medical and family history records of patients with childhood sarcoma.118 The original description of a kindred with a spectrum of tumors that includes soft tissue sarcomas, osteosarcomas, breast cancer, brain tumors, leukemia, and adrenocortical carcinoma (ACC) has been overwhelmingly substantiated by numerous subsequent studies,119 although other cancers, usually of particularly early age of onset, are also observed.102 Germ line alterations of the p53 tumor suppressor gene are associated with LFS.121,122 These are primarily missense mutations that yield a stabilized mutant protein. The spectrum of mutations of p53 in the germ line is similar to somatic mutations found in a wide variety of tumors. Carriers are heterozygous for the mutation, and in tumors derived from these individuals, the second (wild type) allele is frequently deleted or mutated, leading to functional inactivation.123
Several comprehensive databases document all reported germ line (and somatic) p53 mutations and are of particular value in evaluating novel mutations as well as phenotype-genotype correlations.124 Only 60% to 80% of “classic” LFS families have detectable alterations of the gene. It is not yet determined whether the remainder is associated with the presence of modifier genes, promoter defects yielding abnormalities of p53 expression, or simply the result of weak genotype-phenotype correlations (i.e., the broad clinical definition encompasses families that are not actual members of LFS). Other candidate predisposition genes, such as p16, p15, p21, BRCA1, BRCA2, and PTEN, associated with multisite cancer associations have generally been ruled out as potential targets. The role of the hCHK2 checkpoint kinase as an alternative mechanism for functional inactivation of p53 in LFS has been suggested,125 although its place as a major contributor to the phenotype has been controversial.126
Germ line p53 alterations have also been reported in some patients with cancer phenotypes that resemble the classic LFS phenotype. Between 3% and 10% of children with apparently sporadic RMS or osteosarcoma have been shown to carry germ line p53 mutations.127,128 These patients tend to be younger than those who harbor wild type p53. It appears as well that more than 75% of children with apparently sporadic ACC carry germ line p53 mutations, although in some of these cases, a family history develops that is not substantially distinct from LFS.129,130 These important findings indicate a broader spectrum of patients at risk of germ line p53 mutations, and refined criteria for p53 mutation analysis.131,132 A striking genotype-phenotype correlation has been observed in a unique subgroup of ACC patients in Brazil in whom the same germ line p53 mutation at codon 337 has been observed in 35 unrelated kindred.133 Other cancers typical of LFS are not observed in these families, and the functional integrity of the mutant protein appears to be regulated by alterations in cellular pH,134 which suggests potential biologic mechanisms in ACC cells by which the p53 mutation leads to malignant transformation. All these observations suggest that germ line p53 alterations may be associated with early-onset development of the childhood component tumors of the syndrome.135 It is not clear what clinical significance these findings have in that no studies of prognostic significance or potential impact on anticancer treatment modalities are reported. Nevertheless, in light of the critical role played by p53 in the initiation and potentiation of gamma irradiation or chemotherapy-induced DNA damage repair, studies into the effect of such germ line mutations on the potentiation of tumor development related to therapeutic interventions would be important.
The variability in age of onset and type of cancer among LFS families suggests modifier effects on the underlying mutant p53 genotype. Analysis of mutant genotype-to-phenotype correlations reveals intriguing observations. Nonsense, frameshift, and splice mutations yield a truncated or nonfunctional protein commonly associated with early-onset cancers, particularly brain tumors. Missense mutation in the p53 DNA binding domain are frequently observed in the setting of breast and brain tumors, while adrenocortical cancers are the only group that are associated with mutations in the non-DNA binding loops. Age of onset modifiers have also now been established. The protein murine double-minute-2 (MDM2) is a key negative regulator of p53 and targets p53 toward proteasomal degradation. The MDM2 single nucleotide polymorphism 309 increases Sp1 transcription factor binding, leading to increased MDM2 expression levels. Coinheritance of the MDM2 single nucleotide polymorphism 309 T/G isoform is associated with earlier-onset cancer.136 The earlier age of onset of cancers with subsequent generations in mutant p53 LFS families suggests genetic anticipation. This observation can be partially explained by several molecular mechanisms including accelerated telomere attrition from generation to generation, absence of the PIN3 polymorphism, or excessive DNA copy number variation in p53 mutation carriers, all of which may be useful predictive markers of tumor age of onset.136–138 Thus, although germ line p53 mutations establish the baseline risk of tumor development in LFS, a complex interplay of modifying genetic cofactors likely defines the specific phenotypes of individual patients.
Beckwith-Wiedemann Syndrome
BWS occurs with a frequency of 1 in 13,700 births. More than 450 cases have been documented since the original reported associations of exomphalos, macroglossia, gigantism, and other congenital anomalies. With increasing age, phenotypic features of BWS become less pronounced. Laboratory findings may include, at birth, hypoglycemia (extremely common), polycythemia, hypocalcemia, hypertriglyceridemia, hypercholesterolemia, and high serum α-fetoprotein levels. Early diagnosis of the condition is crucial to avoid deleterious neurologic effects of neonatal hypoglycemia and to initiate an appropriate screening protocol for tumor development.139 The increased risk for tumor formation in BWS patients is estimated at 7.5% and is further increased to 10% if hemihyperplasia is present. Tumors occurring with the highest frequency include Wilms tumor, hepatoblastoma, neuroblastoma, and ACC.20
The genetic basis of BWS is complex. Various 11p15 chromosomal or molecular alterations have been associated with the BWS phenotype and its tumors.140 It is unlikely that a single gene is responsible for the BWS phenotype. Because it appears that abnormalities in the region impact an imprinted domain, it is more likely that normal gene regulation in this part of chromosome 11p15 occurs in a regional manner and may depend on various interdependent factors or genes. These include the paternally expressed genes IGF-2 and KCNQ10T1 and the maternally expressed genes H19, CDKN1C, and KCNQ1. BWS children who develop rhabdomyosarcoma or hepatoblastoma have epigenetic changes in domain 1, whereas those with Wilms tumor have domain 2 changes or uniparental disomy.141
Chromosomal abnormalities associated with BWS are extremely rare, with only 20 cases having been associated with 11p15 translocations or inversions. The chromosomal breakpoint in each of these cases is always found on the maternally derived chromosome 11. This parent-of-origin dependence in BWS suggests that the chromosome translocations disrupt imprinting of a gene in the 11p15 region. On the other hand, BWS-associated 11p15 duplications (approximately 30 reported cases) are always paternally derived, and the duplication breakpoints are heterogeneous.142 Paternal uniparental disomy, in which two alleles are inherited from one parent (the father), has been reported in approximately 15% of sporadic BWS patients.143 It is interesting that the insulin/IGF-2 region is always represented in the uniparental disomy, although the extent of chromosomal involvement is highly variable. Alterations in allele-specific DNA methylation of IGF-2 and H19 reflect this paternal imprinting phenomenon.143 A minority of BWS patients have demonstrable constitutional DNA sequence alterations, the most common of these being CDKN1C mutations.144 Twenty-five percent to 50% of BWS patients exhibit biallelic rather than monoallelic expression of IGF-2. Another 50% have epigenetic mutations resulting in loss of imprinting of KCNQ10T0. Of interest, epigenetic changes, such as methylation and chromatin modification, occur in many pediatric and adult cancers,145 indicating the value of the BWS model in understanding the broad scope of molecular changes in cancer. Despite the associated cytogenetic and molecular findings for some patients, no single diagnostic test exists for BWS. This observation is not unlike that described for LFS, or perhaps for other multisite cancer phenotypes, in which the clarity of the phenotype is often weak, making the genetic link cloudy and the likelihood of multiple pathways to tumor formation strong.
Gorlin Syndrome
Nevoid basal cell carcinoma syndrome, or Gorlin syndrome, is a rare autosomal dominant disorder characterized by multiple basal cell carcinomas, developmental defects including bifid ribs and other spine and rib abnormalities, palmar and plantar pits, odontogenic keratocysts, and generalized overgrowth.146 The Sonic hedgehog (SHH) signaling pathway directs embryonic development of a spectrum of organisms. Gorlin syndrome appears to be caused by germ line mutations of the tumor suppressor gene PTCH, a receptor for SHH.147,148 Medulloblastoma develops in approximately 5% of patients with Gorlin syndrome. Furthermore, approximately 10% of patients diagnosed with medulloblastoma by the age of 2 are found to have other phenotypic features consistent with Gorlin syndrome and also harbor germline PTCH mutations.149 Although Gorlin syndrome develops in individuals with germ line mutations of PTCH, a subset of children with medulloblastoma harbor germ line mutations of another gene, SUFU, in the SHH pathway, with accompanying LOH in the tumors. Of further note, mice with heterozygous PTC deletions develop RMS.151 Although RMS is not associated with Gorlin syndrome, the mouse studies suggest a possible link between PTC signaling and RMS.152
MALIGNANT RHABDOID TUMORS
Malignant rhabdoid tumors are unusual pediatric tumors that occur as primary renal tumors, but have also been described in lung, liver, soft tissues, and the central nervous system, where they are often termed atypical and teratoid rhabdoid tumors.153 Recurrent chromosomal translocations of chromosome 22 involving a breakpoint at 22q11.2, as well as complete or partial monosomy 22, have been observed, strongly suggesting the presence of a tumor suppressor gene in this area. The hSNF5/INI1 gene has been isolated and has been shown to be the target for biallelic, recurrent inactivating mutations.154 The encoded gene product is thought to be involved in chromatin remodeling. Studies have not only demonstrated the presence of inactivating mutations in the majority of malignant rhabdoid tumors (renal or extrarenal) but also in chronic myelogenous leukemia,155 as well as in a wide variety of other childhood and adult-onset malignancies.156 An intriguing feature in some individuals with malignant rhabdoid tumors is the observation of germ line mutations, suggesting that this family of tumors may occur as a result of a primary inherited defect in one allele of the INI1 gene.157 Further studies of the function of this gene will be important in determining its role in tumorigenesis of this wide spectrum of neoplasms.
PREDICTIVE TESTING FOR GERM LINE MUTATIONS AND CHILDHOOD CANCERS
Several important issues have arisen as a result of the identification of germ line mutations of tumor suppressor genes in cancer-prone individuals and families. These include ethical questions of predictive testing in such families and in unaffected relatives and selection of patients to be tested, as well as the development of practical and accurate laboratory techniques, the development of pilot testing programs, and the role of clinical intervention based on test results. This chapter was not meant to discuss these problems in detail, but one would be remiss to ignore their significance.
For several reasons, testing cannot as yet be offered to the general pediatric population, particularly in light of the demonstrably low carrier rate of the abnormal tumor suppressor genes and the general lack of standardized methods of preclinical screening of carriers. Exceptions to these limitations include screening of gene carriers in families with retinoblastoma, BWS, multiple endocrine neoplasia, familial adenomatous polyposis, and von Hippel-Lindau disease. For some of these diseases, clinical surveillance tools are available, whereas for others, risk-reductive surgery has also been shown to be of value.158,159 In general, it has been demonstrated that genetic testing does not lead to clinical levels of anxiety, depression, or other markers of psychological distress in the children who are tested, or their parents.160,161 However, certain circumstances or personality traits are associated with a greater likelihood of an individual experiencing psychological distress after a positive result.160 Parents now routinely discuss the options of prenatal diagnosis and preimplantation genetic diagnosis. Multidisciplinary teams must be engaged to provide parents and families the necessary tools with which to approach these ethically challenging decisions.162,163 The development of screening programs should address aspects of cost, informed consent (particularly where it affects children), socioeconomic impact on the individual tested, consistency in providing results, and counseling. Concerns of risk of employment, health insurance, or life insurance discrimination exist but may be alleviated by congressional legislation to ban such practices.164
MOLECULAR THERAPEUTICS
With the identification of alterations in a variety of molecular signaling pathways, including activated growth factor signaling pathways (e.g., IGF-2) and altered tumor suppressor pathways (e.g., retinoblastoma), it has become increasingly apparent that these alterations may potentially represent the “Achilles’ heel” for these tumors. New agents targeting the tyrosine kinase enzymes that transduce growth factor signals are at various stages of development in early clinical studies. Several IGF-I receptor (IR) antibodies have been tested in ES and rhabdomyosarcoma, and small-molecule IGF-IR kinase inhibitors are entering clinical trials. Several mTOR inhibitors are now approved for treatment of kidney cancer, and these agents are currently being tested in both NF1 as well as in combinations with IGF-IR inhibitors in pediatric sarcomas.
Fusion proteins derived from tumor-specific translocations may themselves represent targets, either as potential neoantigens that could be targeted by cytotoxic T cells or as targets for novel compounds. It is likely that the molecular characterization of pediatric tumors will lead to novel and perhaps more effective treatment approaches in the near future. It is also likely that some of these innovative approaches will at least initially be integrated into standard therapeutic protocols.
Selected References
The full list of references for this chapter appears in the online version.
3. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971;68:820.
8. Cavenee WK, Dryja TP, Phillips RA, et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 1983;305:779.
10. Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986;323:643.
23. Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990;60:509
32. Rivera MN, Kim WJ, Driscoll DR, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 2007;315:642.
40. Viskochil D, Buchberg AM, Xu G, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990;62:187.
41. Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 2009;19:230.
49. Ammoun S, Cunliffe CH, Allen JC, et al. ErbB/HER receptor activation and preclinical efficacy of lapatinib in vestibular schwannoma. Neuro Oncol 2010;12:834.
50. Brodeur GM, Sekhon G, Goldstein MN. Chromosomal aberrations in human neuroblastomas. Cancer 1977;40:2256.
51. Caron H, van Sluis P, de Kraker J, et al. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N Engl J Med 1996;334:225.
58. Schwab M, Alitalo K, Klempnauer KH, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983;305:245.
59. Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 1985; 313:404.
61. Schwab M, Ellison J, Busch M, et al. Enhanced expression of the human gene N-myc consequent to amplification of DNA may contribute to malignant progression of neuroblastoma. Proc Natl Acad Sci U S A 1984;81:4940.
62. Nakagawara A, Arima-Nakagawara M, Scavarda NJ, et al. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med 1993;328:847.
68. Mosse YP, Laduenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008;455:967.
72. Attiyeh EF, London WB, Mosse YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med 2005;353(21):2243.
74. Ohali A, Avigad S, Ash S, et al. Telomere length is a prognostic factor in neuroblastoma. Cancer 2006;107:1391.
77. Whang-Peng J, Triche T, Knutsen T, et al. Chromosome translocation in peripheral neuroepithelioma. N Engl J Med 1984;311:584.
78. Delattre O, Zucman J, Ploustagel B, et al. Gene fusion with an ETS DNA binding domain caused by chromosome translocation in human cancers. Nature 1992;359:162.
87. van Doorninck JA, Ji L, Schaub B, et al. Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol 2010;28:1989.
89. Lessnick SL, Dacwag CS, Golub TR. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell 2002; 1:393.
92. Kinsey M, Smith R, Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol Cancer Res 2006;4:851.
95. Dias P, Parham DM, Shapiro DN, et al. Myogenic regulatory protein (MyoD1) expression in childhood solid tumors: diagnostic utility in rhabdomyosarcoma. Am J Pathol 1990;137:1283.
97. Scrable H, Witte D, Shimada H, et al. Molecular differential pathology of rhabdomyosarcoma. Genes Chromosomes Cancer 1989;1:23.
99. Rainier S, Johnson LA, Dobry CJ, et al. Relaxation of imprinted genes in human cancer. Nature 1993;362:747.
100. Zhan S, Shapiro DN, Helman LJ. Activation of an imprinted allele of the insulin-like growth factor II gene implicated in rhabdomyosarcoma. J Clin Invest 1994; 94:445.
101. Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet 2005;137C:72.
104. Barr FG, Galili N, Holick J, et al. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet 1993;3:113.
106. Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol 2002;20(11):2672.
109. Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev 2004; 18:2614.
117. Taylor JG 6th, Cheuk AT, Tsang PS, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest 2009;119:3395
118. Li FP, Fraumeni JF Jr. Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst 1969;43:1365.
121. Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990;250:1233.
124. Olivier M, Eeles R, Hollstein M, et al. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat 2002;19:607.
132. Tinat J, Bougeard G, Baert-Desurmont S, et al. 2009 version of the Chompret criteria for Li-Fraumeni syndrome. J Clin Oncol 2009;27(26):e108.
133. Ribeiro RC, Sandrini F, Figueiredo B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A 2001;98:9330.
135. Olivier M, Goldgar DE, Sodha N, et al. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 2003;63:6643.
138. Shlien A, Tabori U, Marshall CR, et al. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proc Natl Acad Sci U S A 2008; 105:11264.
142. Henry I, Bonaiti-Pellie C, Chehensse V, et al. Uniparental paternal disomy in a genetic cancer-predisposing syndrome. Nature 1991;351:665.
148. Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996;85:841.
150. Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet 2002;31:306.
154. Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998;394:203.
157. Biegel JA, Zhou JY, Rorke LB, et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 1999;59:74.
162. Lammens C, Bleiker Aaronson N, Aaronson N, et al. Attitudes towards pre-implantation genetic diagnosis for hereditary cancer. Fam Cancer 2009;8(4):457.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


