William A. Craig, David R. Andes Keywords cefaclor; cefadroxil; cefamandole; cefazolin; cefdinir; cefditoren; cefepime; cefixime; cefmetazole; cefonicid; cefoperazone; cefotaxime; cefotetan; cefoxitin; cefpodoxime; cefpirome; cefprozil; ceftaroline; ceftazidime; ceftibuten; ceftizoxime; ceftobiprole; ceftolozane; ceftriaxone; cefuroxime; cefuroxime-axetil; cephalexin; cephalothin; cephapirin; cephradine; loracarbef; moxalactam
Cephalosporins
Although the discovery of the cephalosporin antibiotic class was reported in 1945, it took almost 2 decades for this class to achieve clinical utility. Giuseppe Brotzu is widely credited for discovery of the broad-spectrum inhibitory effects of sewage outflow in Sardinia, Italy.1 Professor Brotzu subsequently isolated the mold Cephalosporin acremonium (now Acremonium chrysogenum) and demonstrated antimicrobial activity of culture filtrates against both gram-positive and gram-negative bacteria. He also demonstrated the in vivo activity of these culture filtrates in animal infection models and in several patients. The filtrate was administered both locally by injection into skin abscesses and systemically for the treatment of brucellosis and typhoid fever.
A decade after the initial discovery, the cephalosporin substances were isolated and identified as fermentation products of the mold.2 Investigators at Oxford, including Florey and Abraham, systematically studied the physical, chemical, and structural characteristics of cephalosporins, as they had for the penicillin class a decade earlier. Three substances—cephalosporin P, N, and C—were identified. Each of the products possessed antimicrobial activity but only cephalosporin C demonstrated activity against both gram-negative and gram-positive bacteria. In addition, it had advantageous stability in the presence of acid and penicillinases.2 Cephalosporin C became the foundation of subsequent drug development.
The first cephalosporin pharmaceutical, cephalothin, was introduced for clinical use in 1964. There are more than 20 cephalosporin antibiotics in use today (17 in the United States). The cephalosporin class is among the most widely prescribed antimicrobial classes because of its broad spectrum of activity, low toxicity, ease of administration, and favorable pharmacokinetic profile.
Chemistry
Most of the available cephalosporins are semisynthetic derivatives of cephalosporin C. The basic structure of the cephem nucleus includes a β-lactam ring fused to a six-member sulfur-containing dihydrothiazine ring (Fig. 21-1). The cephem nucleus is chemically distinct from the penicillin nucleus, which contains a five-member thiazolidine ring. Basic structure numbering of the cephalosporin ring system begins within the dihydrothiazine ring at the sulfur moiety. The starting material used as the nucleus for current cephalosporin development is 7-aminocephalosporanic acid (7-ACA). Attempts to alter the physiochemical and biologic properties of the cephalosporins by chemical side chain modifications were based on successes with similar structural changes at the 6-aminopenicillanic acid side chain of penicillin.3 Chemical modifications of the basic cephem structure by substitution of constituents at positions C1, C3, and C7 led to the various cephalosporin compounds in use today.4,5 Alterations in position C7 and C3 are also commonly referred to as R1 and R2, respectively. In general, changes at R1 affect the microbial spectrum of activity. These modifications often have an impact on the stability of the compound to enzymatic destruction by β-lactamases or on its affinity for the drug target. Modifications at R2 often alter the pharmacology of the compound. Changes in the R2 constituent may influence the ability of the compound to reach certain infection sites, such as the central nervous system, or may simply prolong the elimination half-life of the drug.
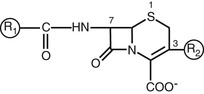
The predominant changes at R1 (position C7) include the addition of an acyl side chain and the substitution of the hydrogen with a methoxy group.5 The R1 methoxy substitution led to the development of the cephamycin group of compounds, including cefoxitin, cefmetazole, and cefotetan. This alteration enhanced resistance to β-lactamase produced by gram-negative anaerobic and aerobic bacteria.5,6 However, these compounds have lower affinity for the penicillin-binding protein (PBP) target in gram-positive bacteria.7 The cephamycin group is structurally related to the cephalosporins but originated as a metabolite from Streptomyces lactamdurans.6 The basic building block of the cephamycin group is cephamycin C. Hydrolysis of cephamycin C produces the 7-ACA nucleus.
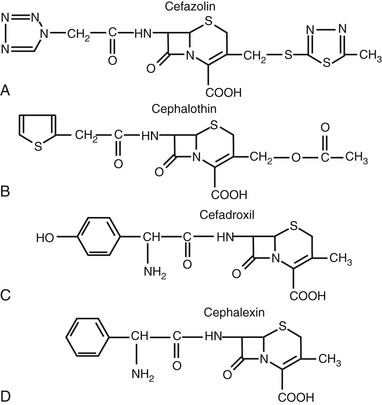
Many modifications of the acyl side chain have been undertaken. The first compounds resulting from addition of a thienyl ring or a tetrazole structure at R1 included the first-generation cephalosporins—cephalothin, cephaloridine, and cefazolin (Fig. 21-2). The simple substitution of an aminobenzyl group in the C7 position is important for oral absorption of the cephalosporins.5 Cephalexin, cephradine, cefaclor, cefprozil, and loracarbef all have this structure or a closely related one (Fig. 21-3). Absorption of later-generation cephalosporins is enhanced by the production of ester formulations. Axetil, proxetil, or pivoxil esters of cefuroxime, cefpodoxime, and cefditoren are currently available.
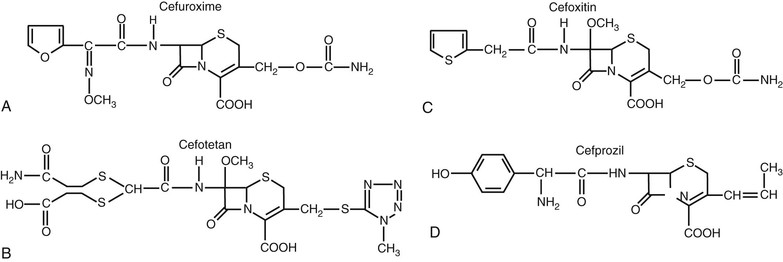
Most of the chemical modifications in cephalosporin development that have resulted in changes in microbiologic spectrum are alterations at the α-carbon of the acyl side chain.5 These changes have ranged from the relatively simple addition of a hydroxyl group to the addition of large synthetic moieties. Each of the acyl side chain alterations has led to enhanced gram-negative potency because of improved β-lactamase stability. The addition of a hydroxyl group at the α-carbon led to the second-generation cephalosporin, cefamandole. The second-generation cephalosporin cefuroxime resulted from the addition of a methoxyimino group in the α position, along with a furyl ring at the β-acyl side chain. Addition of a 2-aminothiazol group to the C7 β-acyl side chain and a methoxyimino group to the α-carbon led to many of the third- and fourth-generation cephalosporins (Figs. 21-4 and 21-5).8,9 Cefotaxime, ceftizoxime, ceftriaxone, cefepime, cefpirome, and cefpodoxime all have a similar structure at the C7 position. Ceftazidime and ceftolozane differ from these drugs in that the methoxyimino group is replaced with a dimethylacetic acid moiety attached to the imino group.8,10 This alteration enhances activity against Pseudomonas aeruginosa but reduces activity against staphylococci. Two other modifications that have resulted in compounds with increased activity against P. aeruginosa are the addition of an ureido-2,3-dioxopiperazine group or a carboxyl group on the α-carbon, producing cefoperazone and moxalactam, respectively.5 These changes are similar to those of piperacillin and carbenicillin.
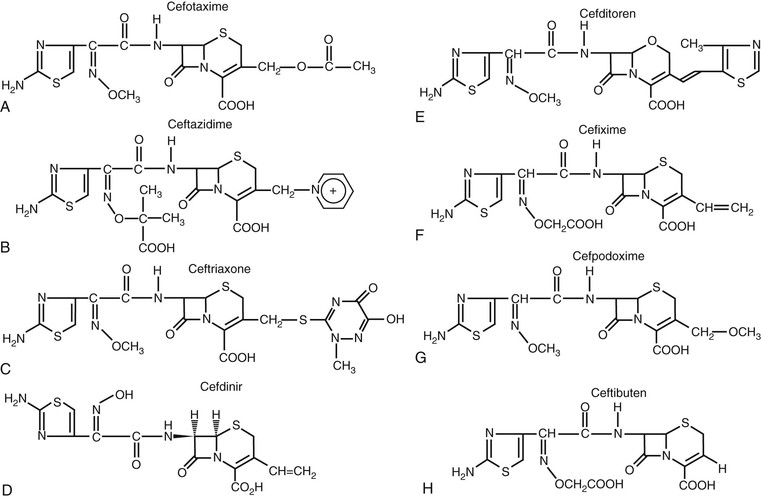
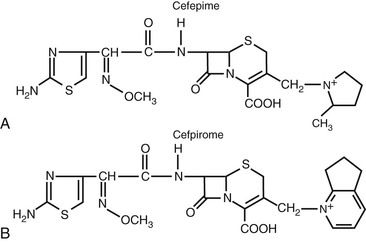
Numerous modifications at R2 (the C3 position) have also played a significant role in the development of the current cephalosporins. An acetoxy side chain is present in cephalothin, cephapirin, and cefotaxime.5 Cephalosporins with this structure can be metabolized in both serum and liver to a less active desacetyl derivative. Such drugs also tend to have a short half-life. A chloride substitution at R2 enhanced the gram-negative spectrum of activity and led to the development of cefaclor, an early second-generation cephalosporin. The unique pharmacology of ceftriaxone results from an R2 modification. Substitution of a heterocyclic thiomethyl group at the C3 position increases biliary secretion and remarkably prolongs the elimination half-life of the compound because of high protein binding.5,11 The addition of a positively charged quaternary ammonium moiety in the C3 position contributed to the development of the fourth-generation cephalosporins, cefepime and cefpirome.9 The chemical modification produces a zwitterion, which enhances the ability of the compound to penetrate the outer membrane of gram-negative organisms. The inclusion of additional amino groups to this ammonium moiety further enhances activity against P. aeruginosa.10 Not all modifications have led to desired effects. The placement of a thiomethyl tetrazole ring (MTT) at the R2 position enhanced antibacterial activity but also resulted in two important adverse effects that have limited use of these compounds.12,13 Cefamandole, cefotetan, cefoperazone, and moxifloxacin (an oxycephem) contain this MTT side chain, which is responsible for coagulation abnormalities related to antagonism of vitamin K action. This side chain is also responsible for the disulfiram-like properties of these compounds.
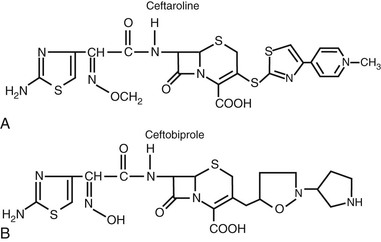
More recently, cephalosporins with enhanced activity against methicillin-resistant Staphylococcus aureus (MRSA) have been developed (Fig. 21-6). A variety of structural alterations at the C3 and C7 positions have increased the drug’s stability to β-lactamase inactivation and enhanced tight binding to the altered penicillin-binding protein 2a′.14,15 Because some of these compounds require more lipophilicity at the C3 position for activity, prodrugs have been required to enhance aqueous solubility.16,17
Classification
There are several microbiologic and pharmacologic differences that could serve as a basis for classification among the cephalosporin drug class. The most widely accepted classification includes five divisions, or generations, based loosely on the microbial spectrum of activity (Table 21-1). The first-generation cephalosporins exhibit activity focused primarily on gram-positive bacteria. The second-generation drugs have enhanced activity against gram-negative bacilli but maintain varying degrees of activity against gram-positive cocci. The cephamycin group is included in the second-generation classification. The cephamycins are noted for their additional activity against gram-negative anaerobic bacteria, such as Bacteroides spp.
TABLE 21-1
Classification of Parenteral and Oral Cephalosporins
| FIRST GENERATION | SECOND GENERATION | CEPHAMYCINS | THIRD GENERATION | FOURTH GENERATION | MRSA-ACTIVE |
| Parenteral Cephalosporins | |||||
| Cefazolin (Ancef, Kefzol) Cephalothin (Keflin, Seffin)* Cephapirin (Cefadyl)* Cephradine (Velosef)* | Cefamandole (Mandol)* Cefonicid (Monocid)* Cefuroxime (Kefurox, Zinacef) | Cefmetazole (Zefazone)* Cefotetan (Cefotan) Cefoxitin (Mefoxin) | Cefoperazone (Cefobid)* Cefotaxime (Claforan) Ceftazidime (Fortaz) Ceftizoxime (Cefizox)* Ceftriaxone (Rocephin) Moxalactam* | Cefepime (Maxipime) Cefpirome (Cefrom)* | Ceftaroline (Teflaro) Ceftobiprole (Zeftera)* |
| Oral Cephalosporins | |||||
| Cefadroxil (Duricef, Ultracef) Cephalexin (Keflex, Biocef, Keftab) Cephradine (Velosef)* | Cefaclor (Ceclor)* Cefprozil (Cefzil) Cefuroxime-axetil (Ceftin) Loracarbef (Lorabid)* | Cefdinir (Omnicef) Cefditoren (Spectracef) Cefixime (Suprax) Cefpodoxime (Vantin) Ceftibuten (Cedax) | |||
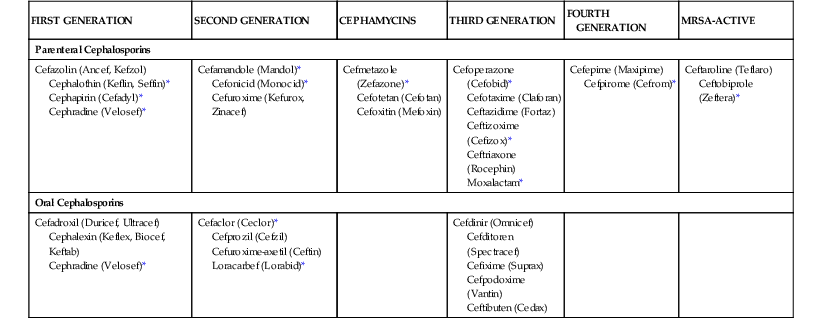
* No longer marketed in the United States.
The third-generation cephalosporins have markedly increased potency against gram-negative bacilli; however, for some compounds in this group, activity against gram-positive cocci is reduced. Among the third-generation group, a few compounds, such as ceftazidime and ceftolozane, are considered separately for activity against P. aeruginosa. The fourth generation has the widest spectrum of activity of the five groups. These drugs, such as cefepime and cefpirome, have activity against most gram-negative bacilli, including P. aeruginosa, and maintain their potency against gram-positive cocci. The third- and fourth-generation drugs combined are also called the extended-spectrum cephalosporins.
The fifth group is referred to as the MRSA-active cephalosporins and currently includes ceftaroline and ceftobiprole (not yet available in the United States). In addition to this unique activity against MRSA among the cephalosporins, these drugs also have enhanced activity against Streptococcus pneumoniae and Enterococcus faecalis. Their activity against gram-negative bacilli is similar to that of the third-generation cephalosporins. Ceftobiprole also has activity against P. aeruginosa.
More recently, a few cephalosporins susceptible to inactivation by extended-spectrum β-lactamases (ESBLs) have been combined with β-lactamase inhibitors to expand their activity against gram-negative bacilli. Ceftazidime and ceftaroline are combined with avibactam, whereas ceftolozane is combined with tazobactam.18
Mechanism of Action
The mechanism of antibacterial activity of cephalosporins is similar to that of other β-lactam drugs. Bacterial growth is inhibited by interference with the synthesis of the cell wall. The primary target of these compounds within the cell wall is the peptidoglycan cross-linkage structure.19 Peptidoglycans are polysaccharide chains consisting of alternating N-acetylglucosamine and N-acetylmuramic acid residues. The polysaccharide chains are cross-linked at the pentapeptide side chain of the N-acetylmuramic acid residues to form a netlike structure. These structures are inserted into the cytoplasmic membrane from the cytoplasm by the action of a group of enzymes that includes transpeptidases, carboxypeptidases, and endopeptidases. The lactam ring provides for penicillins and cephalosporins a conformation similar to the terminal d-alanine-d-alanine of the pentapeptide.20 The antibiotics bind covalently to these enzymes, in particular to the transpeptidases, resulting in loss of enzyme activity.19 The enzyme drug targets are referred to as penicillin-binding proteins (PBPs).21,22
The location of the PBPs relative to the extracellular space differs between gram-positive and gram-negative bacteria. The peptidoglycan of gram-positive bacteria is located on the outer surface of the cell. A complex lipopolysaccharide (LPS) structure is located on the outermost surface of gram-negative bacteria; cephalosporins must first penetrate or diffuse across the LPS membrane to reach the PBPs. The PBP targets within bacteria vary by type and amount. These targets are numbered by convention on the basis of molecular weight, with letters differentiating proteins of similar molecular weight. Gram-positive and gram-negative cocci typically have 3 to 5 PBPs; gram-negative bacilli usually contain 7 to 10 PBPs. The cephalosporin drugs can vary in their affinity for each of these drug targets. At low concentrations, cephalosporins preferentially bind to PBP 3 in gram-negative bacilli, resulting in filament formation with septae.15,23 The events after the covalent binding of cephalosporins to the PBP targets that lead to cell lysis and death are not entirely understood.
In general, cephalosporins are considered to be bactericidal drugs. The rate of killing of bacteria by cephalosporins exhibits minimal dependence on the concentration of the antibiotic.24 Maximal bacterial killing is observed at concentrations four times the minimal inhibitory concentration (MIC). Cephalosporins produce persistent suppression of bacterial growth (i.e., the postantibiotic effect) of several hours’ duration with gram-positive bacteria, but they induce very short or no postantibiotic effects with gram-negative bacilli.25,26 The duration of time during which drug concentrations exceed the MIC is the major determinant of the antibacterial activity of the cephalosporins.26–28
Spectrum of Activity
The cephalosporins are active against a wide variety of aerobic and anaerobic bacteria (Tables 21-2 and 21-3).29–63 Most drugs are active against streptococci and staphylococci. Some of the differences in potency among the agents are magnified by their activity against penicillin-resistant pneumococci. Ceftaroline and ceftobiprole have the greatest potency against this organism, followed by cefditoren, ceftriaxone, cefotaxime, cefepime, and cefpirome.59,63 The cephamycins, ceftazidime, cefixime, and ceftibuten have the poorest activity against methicillin-susceptible staphylococci. Although MRSAs are resistant to all the earlier cephalosporins, the new MRSA-active cephalosporins, ceftaroline and ceftobiprole, exhibit MICs of approximately 0.5 to 4 µµg/mL with such strains.59,63 Enterococci have also consistently been resistant to the cephalosporins, with most MICs greater than 32 µg/mL. However, the new MRSA-active cephalosporins have much lower MICs for ampicillin-susceptible strains. These have ranged from 0.12 to 4 µg/mL for both drugs.59,63
TABLE 21-2
In Vitro Activity of Cephalosporins against Selected Gram-Positive Cocci, Haemophilus influenzae, Moraxella catarrhalis, and Neisseria Species*
| CEPHALOSPORIN | STREPTOCOCCUS PNEUMONIAE | STREPTOCOCCUS PYOGENES | STREPTOCOCCUS AGALACTIAE | VIRIDANS STREPTOCOCCI GROUP | STAPHYLOCOCCUS AUREUS | STAPHYLOCOCCUS EPIDERMIDIS | HAEMOPHILUS INFLUENZAE | MORAXELLA CATARRHALIS | NEISSERIA MENINGITIDIS | NEISSERIA GONORRHOEAE | ||
| (PSSP) | (PRSP) | (MSSA) | (MRSA) | |||||||||
| First Generation | ||||||||||||
| Cefazolin | 0.5/4 | 32/>32 | 0.12/0.12 | 0.12/0.12 | 0.12/0.12 | 0.5/2 | R | 0.5/>32 | 4/16 | 2/4 | — | 16/32 |
| Cephalothin | 0.12/0.25 | 8/16 | 0.05/0.10 | 0.12/0.5 | 0.25/0.50 | 0.12/0.5 | R | 0.5/32 | 4/8 | 4/8 | 0.25/0.5 | 8/32 |
| Cefadroxil (O) | 2/4 | >32 | 0.12/0.25 | 0.25/2 | — | 2/8 | R | 4/>32 | 16/>32 | 2/4 | — | 8/64 |
| Cephalexin (O) | 1/2 | >32 | 0.25/2 | 0.5/4 | — | 2/4 | R | 1/>32 | 8/16 | 2/8 | 2/2 | 4/16 |
| Cephradine (O) | 2/4 | >32 | 0.25/2 | 0.5/2 | — | 1/4 | R | 4/>32 | 4/16 | 2/4 | — | 8/16 |
| Second Generation | ||||||||||||
| Cefamandole | 0.12/0.5 | 8/>32 | 0.12/0.12 | 0.12/0.5 | 0.12/4 | 1/1 | R | 0.5/>32 | 2/8 | 1/4 | 0.12/0.12 | 0.25/4 |
| Cefonicid | 0.5/1 | 4/>32 | 0.12/0.12 | 0.12/2 | 0.12/8 | 1/2 | R | 2/>32 | 0.5/1 | 1/4 | 0.12/0.12 | 0.06/0.5 |
| Cefuroxime | 0.12/0.25 | 4/>32 | 0.12/0.12 | 0.12/0.12 | 0.12/0.5 | 1/2 | R | 0.5/>32 | 1/2 | 0.5/2 | 0.12/0.12 | 0.015/0.25 |
| Cefaclor (O) | 0.5/1 | 16/>32 | 0.06/0.5 | 0.5/2 | — | 1/8 | R | 1/>32 | 2/32 | 0.5/2 | 0.06/0.25 | 0.25/16 |
| Cefprozil (O) | 0.12/0.5 | 8/>32 | 0.03/0.12 | 0.06/0.25 | — | 0.5/2 | R | 0.25/32 | 2/16 | 1/8 | — | 0.12/4 |
| Loracarbef (O) | 0.5/2 | >32 | 0.5/2 | 0.5/2 | — | 1/4 | R | 4/>32 | 1/4 | 0.5/4 | 0.12/0.25 | 0.5/4 |
| Cephamycins | ||||||||||||
| Cefmetazole | 2/16 | >32 | 0.5/0.5 | 2/2 | 2/4 | 4/16 | R | 8/>32 | 1/4 | 0.12/0.5 | 0.12/0.12 | 0.25/4 |
| Cefotetan | 8/16 | >32 | 2/4 | 4/8 | 2/8 | 8/16 | R | 32/>32 | 1/2 | 0.12/2 | 0.12/0.25 | 0.25/2 |
| Cefoxitin | 2/4 | 32/>32 | 1/2 | 2/2 | 4/16 | 4/8 | R | 2/>32 | 1/4 | 0.25/0.5 | 0.12/0.25 | 0.25/4 |
| Third Generation | ||||||||||||
| Cefoperazone | 0.06/0.12 | 4/16 | 0.12/0.12 | 0.12/0.25 | 0.5/1 | 2/4 | R | 2/>32 | 0.015/0.25 | 0.12/2 | 0.12/0.5 | 0.03/0.06 |
| Cefotaxime | 0.015/0.06 | 0.5/2 | 0.015/0.015 | 0.03/0.25 | 0.06/0.25 | 2/2 | R | 4/>32 | 0.008/0.015 | 0.5/1 | 0.004/0.008 | 0.004/0.008 |
| Ceftazidime | 0. 25/1 | 16/>32 | 0.12/0.25 | 0.25/0.5 | 1/2 | 8/32 | R | 8/>32 | 0.06/0.12 | 0.03/0.5 | 0.015/0.06 | 0.03/0.06 |
| Ceftizoxime | 0.25/1 | 16/32 | 0.015/0.015 | 0.12/0.12 | 0.25/2 | 4/8 | R | 4/>32 | 0.015/0.03 | 0.03/0.5 | 0.008/0.03 | 0.008/0.015 |
| Ceftriaxone | 0.03/0.06 | 10.5/2 | 0.015/0.03 | 0.03/0.06 | 0.06/0.25 | 2/4 | R | 4/>32 | 0.008/0.015 | 0.25/0.5 | 0.008/0.015 | 0.002/0.004 |
| Moxalactam | 1/1 | — | 1/2 | — | — | 8/16 | R | 8/>32 | 0.03/0.12 | 0.03/0.12 | 0.008/0.06 | 0.015/0.06 |
| Cefdinir (O) | 0.06/0.12 | 2/8 | 0.015/0.03 | 0.03/0.06 | — | 0.25/0.5 | R | 0.25/>32 | 0.12/0.5 | 0.06/0.25 | 0.06/0.25 | 0.008/0.06 |
| Cefditoren (O) | 0.015/0.03 | 0.5/2 | 0.008/0.015 | 0.06/1 | — | 0.25/1 | R | 0.25/>32 | 0.008/0.015 | 0.25/1 | <0.06/ 0.06 | 0.004/0.06 |
| Cefixime (O) | 0.25/1 | 32/>32 | 0.06/0.25 | 0.12/0.25 | — | 16/>32 | R | 16/>32 | 0.015/0.12 | 0.03/0.5 | <0.06/0.06 | 0.015/0.06 |
| Cefpodoxime (O) | 0.015/0.06 | 2/>32 | 0.06/0.12 | 0.03/0.12 | — | 2/4 | R | 2/>32 | 0.015/0.12 | 1/2 | <0.06/0.06 | 0.06/0.06 |
| Ceftibuten (O) | 4/8 | >32 | 0.5/2 | 4/16 | — | 16/>32 | R | 16/>32 | 0.06/0.12 | 2/4 | <0.06/0.25 | 0.015/0.5 |
| Fourth Generation | ||||||||||||
| Cefepime | 0.06/0.12 | 0.5/2 | 0.015/0.12 | 0.05/0.05 | 0.016/0.03 | 2/4 | R | 2/>32 | 0.06/0.12 | 1/4 | 0.03/0.06 | 0.03/0.06 |
| Cefpirome | 0.03/0.12 | 0.5/2 | 0.008/0.06 | 0.06/0.06 | 0.06/0.25 | 1/2 | R | 1/>32 | 0.06/0.12 | 0.5/2 | 0.06/0.06 | .015/0.12 |
| MRSA-Active | ||||||||||||
| Ceftaroline | 0.008/0.015 | 0.12/0.25 | 0.008/0.015 | 0.015/0.03 | 0.008/0.03 | 0.25/0.25 | 0.5/1 | 0.5/0.5 | 1/4 | 0.008/0.03 | 0.002/0.004 | 0.008/0.03 |
| Ceftobiprole | 0.008/0.015 | 0.25/0.25 | 0.008/0.015 | 0.06/0.12 | 0.06/0.12 | 0.25/0.5 | 0.5/2 | 0.5/1 | 0.5/4 | 0.06/0.25 | 0.002/0.004 | 0.03/0.06 |
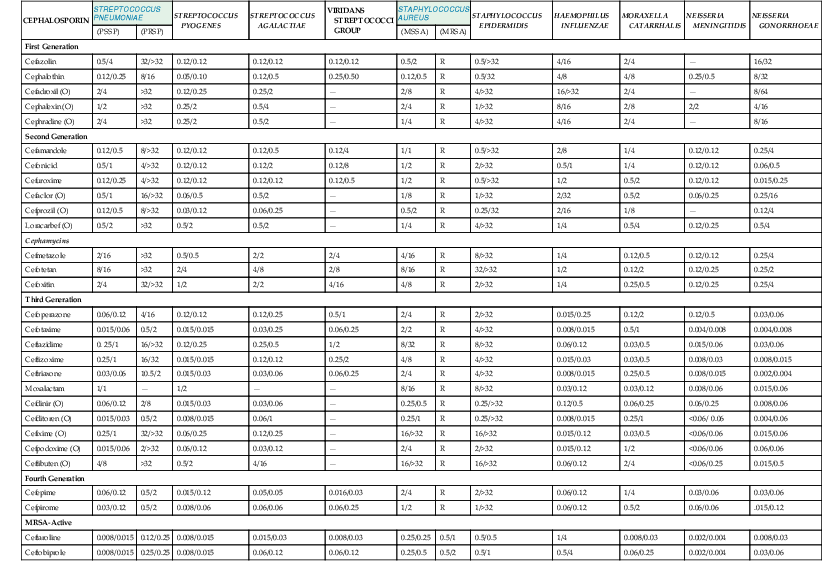
* Minimal inhibitory concentration (MIC) for 50% and 90% of strains in µg/mL.
MRSA, methicillin-resistant Staphylococcus aureus; MSSA, methicillin-susceptible S. aureus; (O), oral; PRSP, penicillin-resistant Streptococcus pneumoniae; PSSP, penicillin-susceptible S. pneumoniae; R, resistant (MICs are variable, but organism is resistant in patients).
TABLE 21-3
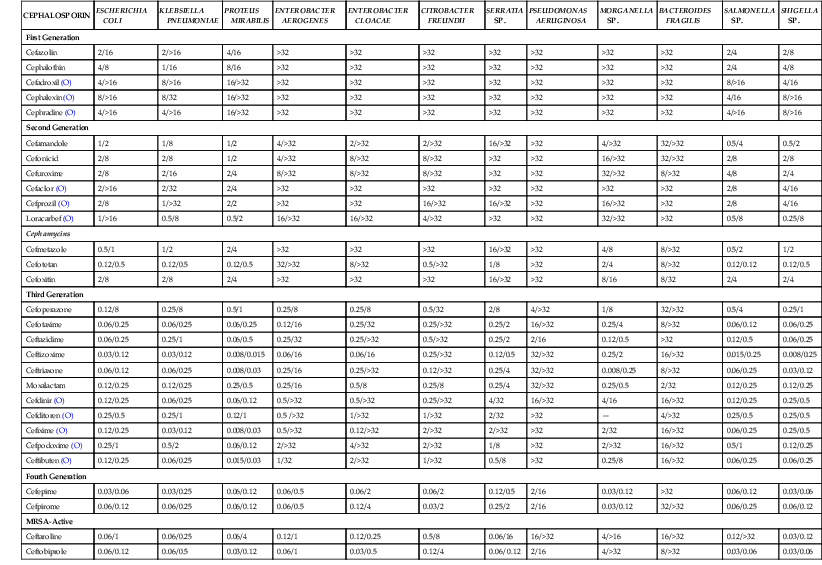
In Vitro Activity of Cephalosporins against Selected Aerobic and Anaerobic Gram-Negative Bacilli*
| CEPHALOSPORIN | ESCHERICHIA COLI | KLEBSIELLA PNEUMONIAE | PROTEUS MIRABILIS | ENTEROBACTER AEROGENES | ENTEROBACTER CLOACAE | CITROBACTER FREUNDII | SERRATIA SP. | PSEUDOMONAS AERUGINOSA | MORGANELLA SP. | BACTEROIDES FRAGILIS | SALMONELLA SP. | SHIGELLA SP. |
| First Generation | ||||||||||||
| Cefazolin | 2/16 | 2/>16 | 4/16 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | 2/4 | 2/8 |
| Cephalothin | 4/8 | 1/16 | 8/16 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | 2/4 | 4/8 |
| Cefadroxil (O) | 4/>16 | 8/>16 | 16/>32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | 8/>16 | 4/16 |
| Cephalexin (O) | 8/>16 | 8/32 | 16/>32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | 4/16 | 8/>16 |
| Cephradine (O) | 4/>16 | 4/>16 | 16/>32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | 4/>16 | 8/>16 |
| Second Generation | ||||||||||||
| Cefamandole | 1/2 | 1/8 | 1/2 | 4/>32 | 2/>32 | 2/>32 | 16/>32 | >32 | 4/>32 | 32/>32 | 0.5/4 | 0.5/2 |
| Cefonicid | 2/8 | 2/8 | 1/2 | 4/>32 | 8/>32 | 8/>32 | >32 | >32 | 16/>32 | 32/>32 | 2/8 | 2/8 |
| Cefuroxime | 2/8 | 2/16 | 2/4 | 8/>32 | 8/>32 | 8/>32 | >32 | >32 | 32/>32 | 8/>32 | 4/8 | 2/4 |
| Cefaclor (O) | 2/>16 | 2/32 | 2/4 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | 2/8 | 4/16 |
| Cefprozil (O) | 2/8 | 1/>32 | 2/2 | >32 | >32 | 16/>32 | 16/>32 | >32 | 16/>32 | >32 | 2/8 | 4/16 |
| Loracarbef (O) | 1/>16 | 0.5/8 | 0.5/2 | 16/>32 | 16/>32 | 4/>32 | >32 | >32 | 32/>32 | >32 | 0.5/8 | 0.25/8 |
| Cephamycins | ||||||||||||
| Cefmetazole | 0.5/1 | 1/2 | 2/4 | >32 | >32 | >32 | 16/>32 | >32 | 4/8 | 8/>32 | 0.5/2 | 1/2 |
| Cefotetan | 0.12/0.5 | 0.12/0.5 | 0.12/0.5 | 32/>32 | 8/>32 | 0.5/>32 | 1/8 | >32 | 2/4 | 8/>32 | 0.12/0.12 | 0.12/0.5 |
| Cefoxitin | 2/8 | 2/8 | 2/4 | >32 | >32 | >32 | 16/>32 | >32 | 8/16 | 8/32 | 2/4 | 2/4 |
| Third Generation | ||||||||||||
| Cefoperazone | 0.12/8 | 0.25/8 | 0.5/1 | 0.25/8 | 0.25/8 | 0.5/32 | 2/8 | 4/>32 | 1/8 | 32/>32 | 0.5/4 | 0.25/1 |
| Cefotaxime | 0.06/0.25 | 0.06/0.25 | 0.06/0.25 | 0.12/16 | 0.25/32 | 0.25/>32 | 0.25/2 | 16/>32 | 0.25/4 | 8/>32 | 0.06/0.12 | 0.06/0.25 |
| Ceftazidime | 0.06/0.25 | 0.25/1 | 0.06/0.5 | 0.25/32 | 0.25/>32 | 0.5/>32 | 0.25/2 | 2/16 | 0.12/0.5 | >32 | 0.12/0.5 | 0.06/0.25 |
| Ceftizoxime | 0.03/0.12 | 0.03/0.12 | 0.008/0.015 | 0.06/16 | 0.06/16 | 0.25/>32 | 0.12/0.5 | 32/>32 | 0.25/2 | 16/>32 | 0.015/0.25 | 0.008/0.25 |
| Ceftriaxone | 0.06/0.12 | 0.06/0.25 | 0.008/0.03 | 0.25/16 | 0.25/>32 | 0.12/>32 | 0.25/4 | 32/>32 | 0.008/0.25 | 8/>32 | 0.06/0.25 | 0.03/0.12 |
| Moxalactam | 0.12/0.25 | 0.12/0.25 | 0.25/0.5 | 0.25/16 | 0.5/8 | 0.25/8 | 0.25/4 | 32/>32 | 0.25/0.5 | 2/32 | 0.12/0.25 | 0.12/0.25 |
| Cefdinir (O) | 0.12/0.25 | 0.06/0.25 | 0.06/0.12 | 0.5/>32 | 0.5/>32 | 0.25/>32 | 4/32 | 16/>32 | 4/16 | 16/>32 | 0.12/0.25 | 0.25/0.5 |
| Cefditoren (O) | 0.25/0.5 | 0.25/1 | 0.12/1 | 0.5 />32 | 1/>32 | 1/>32 | 2/32 | >32 | — | 4/>32 | 0.25/0.5 | 0.25/0.5 |
| Cefixime (O) | 0.12/0.25 | 0.03/0.12 | 0.008/0.03 | 0.5/>32 | 0.12/>32 | 2/>32 | 2/>32 | >32 | 2/32 | 16/>32 | 0.06/0.25 | 0.25/0.5 |
| Cefpodoxime (O) | 0.25/1 | 0.5/2 | 0.06/0.12 | 2/>32 | 4/>32 | 2/>32 | 1/8 | >32 | 2/>32 | 16/>32 | 0.5/1 | 0.12/0.25 |
| Ceftibuten (O) | 0.12/0.25 | 0.06/0.25 | 0.015/0.03 | 1/32 | 2/>32 | 1/>32 | 0.5/8 | >32 | 0.25/8 | 16/>32 | 0.06/0.25 | 0.06/0.25 |
| Fourth Generation | ||||||||||||
| Cefepime | 0.03/0.06 | 0.03/0.25 | 0.06/0.12 | 0.06/0.5 | 0.06/2 | 0.06/2 | 0.12/0.5 | 2/16 | 0.03/0.12 | >32 | 0.06/0.12 | 0.03/0.06 |
| Cefpirome | 0.06/0.12 | 0.06/0.25 | 0.06/0.12 | 0.06/0.5 | 0.12/4 | 0.03/2 | 0.25/2 | 2/16 | 0.03/0.12 | 32/>32 | 0.06/0.25 | 0.06/0.12 |
| MRSA-Active | ||||||||||||
| Ceftaroline | 0.06/1 | 0.06/0.25 | 0.06/4 | 0.12/1 | 0.12/0.25 | 0.5/8 | 0.06/16 | 16/>32 | 4/>16 | 16/>32 | 0.12/>32 | 0.03/0.12 |
| Ceftobiprole | 0.06/0.12 | 0.06/0.5 | 0.03/0.12 | 0.06/1 | 0.03/0.5 | 0.12/4 | 0.06/ 0.12 | 2/16 | 4/>32 | 8/>32 | 0.03/0.06 | 0.03/0.06 |
* Minimal inhibitory concentration (MIC) for 50% and 90% of strains in µg/mL.
MRSA, methicillin-resistant Staphylococcus aureus; (O), oral.
The first-generation cephalosporins are not very active against Haemophilus influenzae or Moraxella catarrhalis. The second-generation drugs are about fourfold more potent against these respiratory pathogens. The third-generation cephalosporins have the lowest MICs for H. influenzae and M. catarrhalis—10 to 100 times lower than those of the second-generation drugs. The first-generation cephalosporins also are not as active against Neisseria spp. as the second-, third-, and fourth-generation drugs.
Although all of the cephalosporins are considered to be active against Escherichia coli, Klebsiella pneumoniae, and Proteus mirabilis, the potency of the third- and fourth-generation drugs and the MRSA-active cephalosporins is 10- to 100-fold greater than that of the first- and second-generation cephalosporins. The increased potency of the later-generation drugs extends to strains of Enterobacter, Serratia, Citrobacter, and Morganella spp., which are usually resistant to the first- and second-generation drugs. Several cephalosporins, such as ceftazidime, ceftolozane, cefoperazone, cefepime, cefpirome, and ceftobiprole, are active against many strains of P. aeruginosa. The third- and fourth-generation cephalosporins also exhibit enhanced potency against strains of Salmonella and Shigella.
Many cephalosporins are active against penicillin-susceptible gram-positive anaerobes, such as peptostreptococci.64,65 Among the cephalosporins, the best activity against Bacteroides fragilis is with drugs such as the cephamycins—cefotaxime, ceftriaxone, and ceftizoxime. Many of the drugs are active against spirochetes, including the agents of Lyme disease and syphilis.66,67As a group, the cephalosporins have very poor activity against Chlamydia, Mycoplasma, and Listeria spp.68–70
Mechanisms of Resistance
Four general mechanisms can result in resistance to cephalosporin antibiotics: (1) antibiotic destruction by hydrolyzing β-lactamase enzymes, (2) reduced penetration of the antibiotic through the LPS membrane to the PBP target, (3) enhanced efflux of the drug from the periplasmic space, and (4) alteration in the PBP target, resulting in reduced binding affinity. Most often, resistance in a bacterial population is caused by a single mechanism; however, an increasing percentage of organisms are exhibiting multiple mechanisms.71
Production of β-lactamase enzymes that hydrolyze the β-lactam ring is a predominant resistance mechanism for many gram-negative bacteria. With staphylococci, most cephalosporins, with the exception of cephaloridine, are poorly hydrolyzed by staphylococcal penicillinases. Resistance to cephalosporins in these organisms is due almost entirely to reduced binding affinity of the PBPs. Although all gram-negative bacilli produce β-lactamase enzymes, the type and amount of enzyme varies among organisms. These enzymes are located in the periplasmic space between the outer LPS membrane and the inner cell membrane. Drugs that are able to penetrate the outer membrane can be degraded before reaching the PBP target. The net antimicrobial activity of cephalosporins against gram-negative bacilli is dependent on the rate of penetration across the outer membrane and the stability of the drug to the various hydrolyzing β-lactamases. The penetration of drugs across the outer membrane is through water-filled channels formed by various membrane proteins, termed porins. Movement through porin channels depends on the size, shape, charge, and hydrophilic properties of the compound. The concentration of a drug with a slow rate of penetration is low relative to the amount of β-lactamase within the periplasmic space, so the relatively high β-lactamase concentration can enhance antibiotic inactivation. For a drug that can penetrate rapidly, the converse is true. For example, the zwitterion of cefepime enhances movement across the membrane, resulting in high concentrations in the periplasmic space and a relative net resistance to drug hydrolysis.72
The number of unique β-lactamases is markedly increasing with more than 1000 distinct enzymes. Some of this increase is due to point mutations in the β-lactamases. These proteases may be genetically encoded chromosomally or extrachromosomally. Stable derepression of a chromosomal mutation is a common genotypic scenario. This is observed predominantly in Enterobacter spp., Serratia spp., Citrobacter freundii, and P. aeruginosa.73 Transmissible plasmids have acquired the genes for AmpC enzymes and are appearing in bacteria, such as E. coli and K. pneumoniae, that lack or poorly express a chromosomal enzyme.74 The AmpC cephalosporinase is capable of inactivating almost all current cephalosporins, including the cephamycins. Emergence of this type of resistance is frequent when infections resulting from these organisms are treated only with broad-spectrum cephalosporins.75,76
Many of the newer enzymes represent variants of the common plasmid-encoded Temoneira (TEM)-1, TEM-2 and sulfhydryl variable (SHV)-1 β-lactamases. These enzymes have been observed most commonly in K. pneumoniae and E. coli and are referred to as extended-spectrum β-lactamases (ESBLs) because they are capable of inactivating many third- and fourth-generation cephalosporins.77,78 The new MRSA-active cephalosporins are also susceptible to inactivation by ESBLs.79,80 These enzymes are the result of amino-acid substitutions related to point mutations in the common β-lactamase genes (i.e., TEM and SHV). SHV ESBLs are the most common ESBLs found in K. pneumoniae.81 More recently, the cefotaxime/ceftazidime-hydrolyzing (CTX-M) family of β-lactamases, derived from the chromosomal enzyme of Kluyvera spp., have spread into K. pneumoniae and E. coli.82 This worldwide spread of these organisms is complicating initial treatment of urinary tract infections in many regions.83 Gram-negative bacilli producing carbapenemases are also emerging in certain parts of the world. These enzymes, often called K. pneumoniae carbapenemases (KPCs), are capable of hydrolyzing most of the cephalosporins.84
The different β-lactamase enzymes can vary significantly in their affinity for drugs within the cephalosporin class. For example, cefepime, cefpirome, and ceftobiprole are less susceptible than other cephalosporins to inactivation by AmpC cephalosporinases.79,85,86 The cephamycins are not susceptible to inactivation by ESBLs. Ceftazidime and cefpodoxime generally exhibit high sensitivity to inactivation by ESBLs.77 On the other hand, cefepime, cefpirome, and even cefotaxime and ceftriaxone may exhibit only modest inactivation, and the organism may still be considered susceptible by standard susceptibility testing.
Tests using ceftazidime, cefpodoxime, cefotaxime, or ceftriaxone to identify strains of K. pneumoniae, E. coli, and P. mirabilis producing ESBLs have been devised for clinical laboratories by the Clinical and Laboratory Standards Institute (CLSI).87 However, the identification of ESBLs in the clinical laboratory is complicated by new emerging ESBLs called the CTX-M enzymes, which hydrolyze ceftazidime much less than other third- and fourth-generation cephalosporins.88–90 The previous recommendation of the CLSI was that all such strains should be called resistant to cephalosporins other than the cephamycins, even if they are susceptible on standard susceptibility tests.87 This recommendation was in line with a large, prospective observational study of patients with Klebsiella bacteremia that reported a significantly poorer outcome in patients treated with third- and fourth-generation cephalosporins, compared with carbapenems.91 The poorer outcome was observed primarily in strains with MICs greater than 2 µg/mL. The current susceptibility breakpoints for cephalosporins have been lowered in the United States, as has already been accomplished in Europe.92,93 According to CLSI, strains with ESBLs that have susceptible MICs are considered treatable with cephalosporins at the cited dosages, but there are some recent studies that would support lower MIC susceptibility breakpoints for cefepime. 94,95
It is unlikely that deletion or mutation of porin proteins causes primary resistance to cephalosporins. However, such changes can alter the relationship between the concentrations of drug and β-lactamase in the periplasmic space, resulting in much more hydrolysis of the cephalosporin. For example, strains of K. pneumoniae containing ESBLs have been shown to be resistant to cephamycins because of the lack of an outer membrane porin protein.96 Porin-deficient strains are especially high in Enterobacter aerogenes.97
The endogenous AcrAB multidrug efflux system in E. coli affects the potency of penicillins but has little effect on the activity of cephalosporins.98 However, resistance in P. aeruginosa has been associated with the MexAB-OprM efflux pump for ceftazidime and the MexXY-Opr efflux pump for cefepime and ceftobiprole.99–101 The differences among these organisms in the impact of somewhat similar pumps are probably due to markedly higher outer membrane permeability in E. coli compared with P. aeruginosa.102
Changes in the PBP target are responsible for reduced cephalosporin affinity and subsequent drug resistance for S. pneumoniae, MRSA, H. influenzae, and some Neisseria gonorrhoeae strains. Changes in the PBP target may result from amino-acid substitutions or insertions. Almost 40 amino-acid substitutions in PBP 2b have been described in the pneumococcus.103,104 Multiple mutations have also been described in PBP 2a′ of a strain of MRSA resulting in resistance to ceftobiprole.105 Resistance in S. pneumoniae and several other pathogens has also resulted from the insertion of resistance sequences from other related species.
Pharmacologic Properties
The pharmacologic properties of selected cephalosporins are listed in Table 21-4.106–110,111–132,133–135 Cephalosporins are polar, water-soluble compounds. Within each of the first-, second-, and third-generation classifications, there are both oral and parenteral formulations. The fourth-generation compounds and those with activity against MRSA are available for parenteral use only. The parenteral formulations are available for both intravenous and intramuscular administration. All of the parenteral formulations, with the exception of cephradine, are stable in solution at room temperature for 24 hours or longer.136,137 Drug stability at room temperature facilitates use of these compounds for home intravenous therapy, including continuous infusions. Many of the parenteral compounds can also be administered by the intraperitoneal route for treatment of peritoneal infections associated with continuous ambulatory peritoneal dialysis.138 Formulations of the oral cephalosporins are available as tablets, capsules, or suspensions.
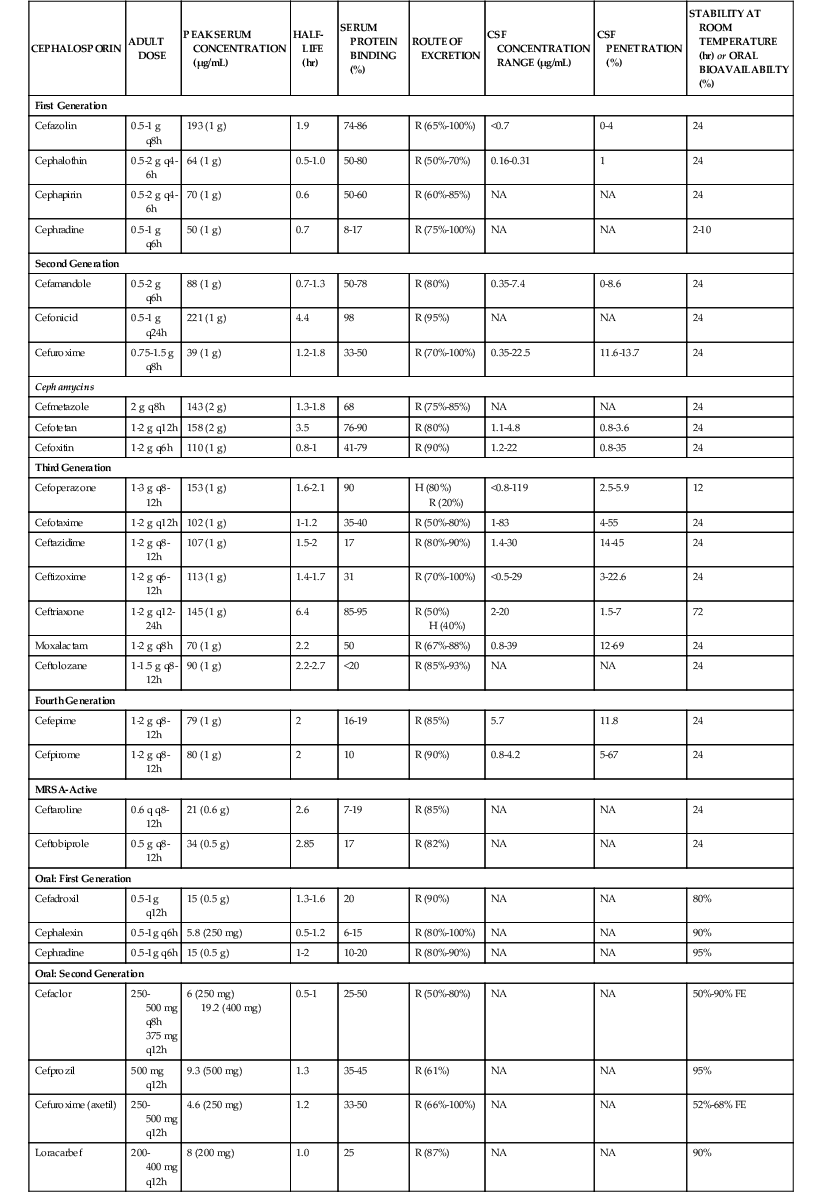
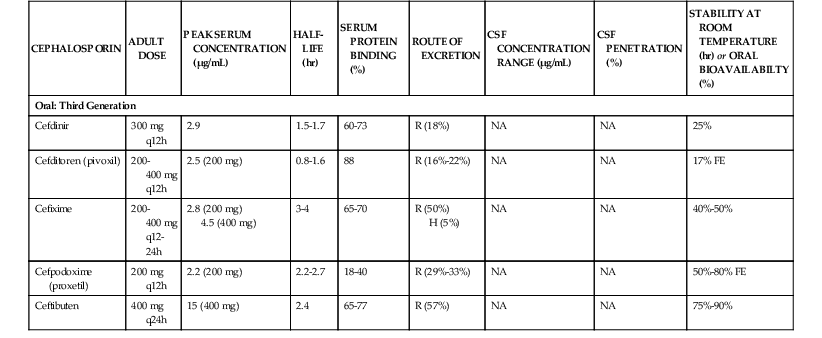
In contrast to many other β-lactams, oral preparations of the cephalosporins are stable in the acid milieu of the upper gastrointestinal tract. Cephalosporins can be actively absorbed if their structure mediates transport by the dipeptide and tripeptide transport systems in the brush border membrane of the small intestine.139 Cephalexin, cephradine, cefadroxil, cefaclor, cefprozil, and loracarbef have an aminobenzyl group or a similar group in the C7 position and have high oral bioavailability (80% to 95%). Ceftibuten, cefixime, and cefdinir have other groups in the C7 position and exhibit more variable bioavailability after oral dosing. Absorption by the dipeptide and tripeptide transport systems appears to be both pH and calcium dependent.139 Drugs that have low oral bioavailability can be esterified to enhance absorption; the ester prodrug is hydrolyzed after absorption in the intestinal epithelial cells. The esters commonly used include axetil, proxetil, and pivoxil formulations.140 Ester prodrug formulations exist for cefuroxime, cefditoren, and cefpodoxime. Absorption of the ester is still not complete; in fact, the percent oral bioavailability of ester formulations is lower than that of most nonesterified compounds. Absorption of the ester formulations is enhanced by concomitant food intake140 because food within the stomach delays gastric emptying and prolongs contact with the mucosal surface.
Distribution of cephalosporins within the body is governed by the lipid solubility of the drug and the extent of protein binding. β-Lactams bind almost exclusively to albumin. The extent of protein binding can vary from less than 10% to as much as 98%.126 Because only unbound drug can pass through capillary pores into interstitial fluid or across cell membranes into intracellular fluid, avidly bound compounds tend to exhibit high serum concentrations and low tissue concentrations. In general, the cephalosporins are largely confined to the extracellular compartment. Drug concentrations in subcutaneous blisters, a model for extracellular drug penetration, are similar to those found in serum.126,141 Techniques measuring extracellular drug concentrations in human tissues (e.g., microdialysis) have demonstrated that concentrations of unbound drug in interstitial fluid are also similar to those in serum.142,143 The cephalosporins have relatively poor intracellular concentrations. Tissue homogenates, which mix intracellular and extracellular fluid, always provide concentrations that are lower than those in serum because of dilution by the larger intracellular volume.141 This group of compounds does not achieve intracellular concentrations adequate to treat most intracellular pathogens (e.g., Legionella spp.).144
In the absence of infection, drug concentrations in the cerebrospinal fluid (CSF) and in the vitreous humor are low. None of the oral cephalosporins achieve therapeutic concentrations in the CSF. Penetration of most parenteral drugs from the first- and second-generation groups is similarly poor. Parenteral cefuroxime is an exception, and this drug also has the lowest MICs for common meningeal pathogens among the first- and second-generation cephalosporins.129 The parenteral third- and fourth-generation drugs, such as ceftriaxone, cefotaxime, ceftazidime, and cefepime, achieve concentrations that would allow treatment of central nervous system infections.130–133,134,135 The presence of an active transport system that transports many cephalosporins from the CSF back to serum contributes to the low drug levels in the CSF observed with many of the earlier-generation drugs. The transport protein involved in this system is similar to the protein involved in renal secretion of β-lactam antibiotics.145 Ceftriaxone, cefotaxime, ceftazidime, and cefepime exhibit minimal renal tubular secretion and are poor substrates for the choroid plexus pump, contributing to higher CSF concentrations. Probenecid is a competitive substrate for this pump and can produce higher concentrations with drugs that are effluxed by this transport system.146 Infection results in higher CSF levels because inflammation can enhance penetration as well as interfere with efflux by active transport.
Very few drugs from the cephalosporin class are extensively metabolized. The three exceptions are cefotaxime, cephalothin, and cephapirin, which undergo deacetylation of the acetoxymethyl side chain in the liver.147,148 The metabolic desacetyl products still possess modest microbiologic activity. The elimination half-life of desacetylcefotaxime is significantly longer than that of the parent compound, allowing less frequent administration of this cephalosporin.148 The remaining drugs in the cephalosporin class are excreted from the body unchanged.
Most cephalosporins are eliminated by the kidney, with half-lives of 1 to 2 hours. The major mechanism for renal excretion of many compounds is tubular secretion. This active transport process is largely unaffected by protein binding.129 Probenecid inhibits this organic acid transport system and can prolong the half-life of these compounds. For several compounds, glomerular filtration is more important, and protein binding can significantly prolong their elimination half-life.149 For some drugs, the elimination half-life is 3 to 8 hours, allowing for 12- and 24-hour dosing intervals. A few compounds with high protein binding and high molecular weights, such as ceftriaxone and cefoperazone, are eliminated to a large extent by the biliary route.150 Between 50% and 70% of the active parent compound may be recovered in the bile and eventually in the feces.
The maximal daily doses of agents eliminated primarily by the kidney must be reduced for patients with renal impairment. Most often, this adjustment includes both a reduction in dose level and lengthening of the dosing interval. Recommended dose adjustments for various degrees of renal impairment and for patients receiving dialysis are presented in Table 21-5 for the currently available cephalosporins.151,152 For drugs eliminated by the biliary system, such as ceftriaxone, dose adjustments are unnecessary unless concomitant severe hepatic insufficiency and renal insufficiency are present.151 Most of the cephalosporins excreted by the renal route are eliminated by hemodialysis. Between 20% and 50% of the parent compound is removed after a usual dialysis session. It is recommended that the drug be given again after hemodialysis. On the other hand, few cephalosporins are removed to any significant extent (<10%) by peritoneal dialysis, and additional dosing is not recommended after a peritoneal dialysis session. Continuous venous hemofiltration (CVH) is frequently used in critically ill patients. Compounds eliminated by the kidneys and by hemodialysis are also removed by CVH. Most often, the efficiency of drug removal is thought to be similar to a creatinine clearance of 10 to 30 mL/min, and appropriate dosing modification is recommended.153
TABLE 21-5
Dosing Adjustment of Cephalosporins in Patients with Renal Insufficiency
| CEPHALOSPORIN | USUAL ADULT DOSING REGIMEN | DOSING REGIMEN WITH RENAL INSUFFICIENCY | DOSING REGIMEN WITH DIALYSIS | |||
| GFR <10 mL/min | GFR 10-50 mL/min | GFR 50-90 mL/min | Hemodialysis | CAPD | ||
| First Generation | ||||||
| Cefazolin | 1 g q8h | 0.5-1 g q24h | 0.5-1 g q12h | NC | 0.5-1 g after | 0.5 g q12h |
| Second Generation | ||||||
| Cefuroxime | 1.5 g q8h | 0.75g q24h | 0.75 g q8-12h | NC | 0.75 g after | 0.75 q24h |
| Cephamycins | ||||||
| Cefotetan | 2 g q12h | 1 g q24h | 2 g q24h | NC | 1 g after | 1 g q24h |
| Cefoxitin | 2 g q6h | 1 q q12h | 2 g q12h | 2 g q8h | 1 g after | 1 g q12h |
| Third Generation | ||||||
| Cefotaxime | 2 g q8h | 2 g q24h | 2g q12h | NC | 1 g after | 1 g q24h |
| Ceftazidime | 2 g q8h | 0.5 g q24h | 2g q24h | 2 g q12h | 1 g after | 0.5 q24h |
| Ceftriaxone | 1 g q24h | NC | NC | NC | None | NC |
| Fourth Generation | ||||||
| Cefepime | 2 g q12h | 0.5-1 g q24h | 1g q24h | NC | 1 g after | 0.5-1 g q24h |
| Cefpirome | 2 g q12h | 0.5 g q12h | 1g q12h | NC | 1 g after | 0.5 g three times weekly |
| MRSA-Active* | ||||||
| Ceftobiprole | 0.5 g q8h | 0.25 g q12h | 0.5 g q12h | NC | 0.25 g after | 0.25 g q12h |
| Ceftaroline | 0.6 g q12h | 0.2 g q12h | 0.3-0.4 g q1h | NC | 0.2 g after | 0.2 g q12h |
| Oral: First Generation | ||||||
| Cefadroxil | 500 mg q12h | 500 mg q24h | 500 mg q24h | NC | 500 mg after | 500 mg q24h |
| Cephalexin | 500 mg q6h | 250 mg q12h | 500 mg q12h | NC | 500 mg after | 500 mg q12h |
| Oral: Second Generation | ||||||
| Cefprozil | 500 mg q12h | 250 mg q24h | 500 mg q24h | NC | 500 mg after | 250 mg q24h |
| Cefuroxime (axetil) | 500 mg q8h | 500 mg q24h | 500 mg q12h | NC | 500 mg after | 500 mg q24h |
| Oral: Third Generation | ||||||
| Cefdinir | 300 mg q12h | 300 mg q24h | NC | NC | 300 mg after | 300 mg q24h |
| Cefditoren | 400 mg q12h | 200 mg q24h | 200 mg q12h | NC | None | 200 mg q24h |
| Cefixime | 400 mg q24h | 200 mg q24h | 300 mg q24h | NC | 300 mg after | 200 mg q24h |
| Cefpodoxime | 200 mg q12h | 200 mg q24h | NC | NC | 200 mg after | 200 mg q24h |
| Ceftibuten | 400 mg q24h | 100 mg q24h | 200 mg q24h | NC | 300 mg after | 100 mg q24h |





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


