INTRODUCTION
SUMMARY
Complex feedback pathways regulate the passage of cells through the G1, S, G2, and M phases of the growth cycle. Two key checkpoints control the commitment of cells to replicate DNA synthesis and to mitosis. Many oncogenes and defective tumor-suppressor genes promote malignant change by stimulating cell-cycle entry, or disrupting the checkpoint response to DNA damage. Advances in the understanding of genetic and epigenetic mechanisms of gene regulation provide the basis for novel therapeutic approaches. This chapter presents the pathways and the genetic and epigenetic alterations that regulate cell replication, and highlights the various oncogenes and tumor-suppressor genes that are involved in hematologic malignancies.
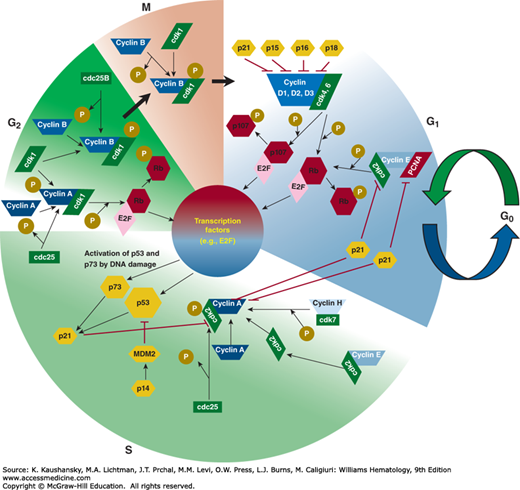
Mitosis is the final step of a defined program—the cell cycle—that can be separated into four phases: the G1, S, G2, and M phases (Fig. 16–1). A number of surveillance systems (checkpoints) control the cell cycle and interrupt its progression when DNA damage occurs or when cells have failed to complete a necessary event.1 These checkpoints have been given an empirical definition: When the occurrence of event B is dependent on the completion of prior event A, the dependence is a result of a checkpoint if a loss-of-function mutation can be found that relieves the dependence.1 Three major cell-cycle checkpoints have been discovered: the DNA damage checkpoint, the replication checkpoint, and the spindle-pole body duplication checkpoint.2,3,4 The functional consequence of failure to “satisfy” the requirements of a cell-cycle checkpoint is usually death by apoptosis. However, small numbers of genetically altered cells may survive. Cells with defective checkpoints have an advantage when selection favors multiple genetic changes. Cancer cells often are missing one or more checkpoints, which facilitates a greater rate of genomic evolution.5
Acronyms and Abbreviations:
ALL, acute lymphoid leukemia; AML, acute myelogenous leukemia; APC, anaphase-promoting complex; APL, acute promyelocytic leukemia; ATM, ataxia-telangiectasia mutated; ATR, ATM and Rad3 related; cdc, cell division cycle; cdk, cyclin-dependent kinase; CDKI, cyclin-dependent kinase inhibitor; Chk, checkpoint kinase; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; CTD, carboxy-terminal domain; DDR, DNA damage response; DSIF, DRB-sensitivity–inducing factor; ER, endoplasmic reticulum; FLAM, flavopiridol, cytarabine, mitoxantrone; GADD, growth arrest and DNA damage; HAT, histone acetyltransferase; HDAC, histone deacetylase; HDACI, histone deacetylase inhibitor; HR, homologous recombination; Id1, inhibitor of DNA-binding 1; INK4, inhibitor of kinase 4; JAK, Janus-associated kinase; MAPK, mitogen-activated protein kinase; MCL, mantle cell lymphoma; MDM2, murine double minute protein 2; MLL, mixed-lineage leukemia; MTA, 5′-deoxy-5′-(methylthio)adenosine; MTAP, methylthioadenosine phosphorylase; NELF, negative elongation factor; N-TEF, negative transcription elongation factor; ODC, ornithine decarboxylase; PDGF, platelet derived growth factor; PI3K, phosphatidylinositol 3′-kinase; PLZF, promyelocytic leukemia Kruppel-like zinc finger; PML, promyelocytic leukemia; P-TEFb, positive transcription elongation factor; RARα, retinoic acid receptor α; RB, retinoblastoma gene; rPTK, receptor protein-tyrosine kinase; STAT, signal transducer and activator of transcription; TGF-β, transforming growth factor-β; TKI, tyrosine kinase inhibitor; UPR, unfolded protein response.
A disturbance of cell-cycle regulation is an important pathway in the development of many hematologic malignancies as a result of mutations in tumor-suppressor genes or oncogenes. Until the end of the 20th century, it was believed that the only mechanism by which the “gatekeepers” of the cell cycle could be inactivated was deletion or mutation (gain-of-function or loss-of-function mutations). Progress in the understanding of the regulation of gene expression put emphasis on another mechanism of gene inactivation, called epigenetic regulation (Chap. 10). This term summarizes several molecular modifications, including histone deacetylation, CpG-island hypermethylation, ubiquitination, and phosphorylation, etc.
CYCLINS AND CYCLIN-DEPENDENT KINASES
Table 16–1 lists Cdks, associated partners, and their functions.
| Cdk | Associated Partner Cyclin | Function |
|---|---|---|
| Cdk1 | Cyclin A, B | G2/M |
| Cdk2 | Cyclin A, D, E; C | G1/S; S; G2/M |
| Cdk3 | Cyclin C | G0 exit |
| Cdk4 | Cyclin D | G1; G1/S |
| Cdk5 | p35, p39 | Neuronal processes (neuron survival/death, migration, cortical layering, synaptic plasticity, etc.) |
| Cdk6 | Cyclin D | G1; G1/S |
| Cdk7 | Cyclin H, Mat1 | Cdk1, 2, 4/6 activation; transcriptional regulation |
| Cdk8 | Cyclin C, MED12, MED13 | Transcriptional regulation |
| Cdk9 | Cyclin T1, T2 | Transcriptional regulation |
| Cdk10 | Ets-2 | G2/M |
| Cdk11 | RanBPM, RNPS1, casein kinase, cyclin L | RNA splicing; transcriptional regulation; apoptosis |
| Cdk12 | Cyclin K, L (?) | Transcriptional regulation; alternative splicing |
| Cdk13 | Cyclin K, L (?) | Transcriptional regulation; alternative splicing |
Early experiments on the control of mitosis in human cells provided evidence for the existence of factors called M-phase and S-phase promoting factors.6 The key element of S-phase promoting factor was thought to be cell division cycle (cdc) 2. Experiments performed in Xenopus eggs showed that cdc2 is an M-phase–specific histone H1 kinase,7 but is just one subunit of a regulatory complex. A second component is cyclin B, which is synthesized in interphase and degraded in mid-mitosis. More than 10 members of the mammalian cyclin family have been identified. Most of these cyclins interact with a group of cdc2-related kinases called cyclin-dependent kinases (cdks),8,9 while others are involved in alternate splicing processes.10 Phosphorylation of tyrosine 15 is the key event in regulating human cdc2 activity. Threonine 14 also is phosphorylated in G2 phase. Dephosphorylation at both phosphorylation sites is required for mitotic initiation. Cdc2 interacts with cyclin B in mitosis, whereas the cdc2/cyclin A complex is formed before mitosis and is required for progression through late G2 phase.11 Thus, cyclins A and B are also called the mitotic cyclins, because they are upregulated in late G2 or G2/M phase and undergo proteolysis in M phase. The exit from mitosis is characterized by the abrupt ubiquitination and subsequent degradation of cyclin B. Cells with a defective cyclin B degradation mechanism or without mitotic cyclin B easily become aneuploid. There is evidence that cyclin A acts at the G2/M transition and binds cdk2 in S phase. Cyclin A is mandatory for the downregulation of anaphase-promoting complex (APC).12 Overexpression of cyclin A in G1 phase leads to an accelerated entry into S phase.13 Because cdc2 is able to interact with mitotic and G1 cyclins, it is likely that one protein kinase potentially can fulfill several different functions in the cell cycle at various checkpoints. Notably, there is increasing evidence that cdc2 is directly involved in regulating the DNA damage response (DDR), including DNA damage checkpoint activation and DNA repair (particularly homologous recombination [HR]).14 There are several cdc2-related protein kinases in humans that interact with the corresponding cyclins. Originally, three cdc2-related proteins were isolated, which were able to replace deficient cdc28 function in budding yeast: cdk1, cdk2, and cdk3.15,16,17 Other cdks are termed cdk4,18 cdk5,19 and cdk6.20 Cdk4/6 has been in the focus of tumor-suppressor gene research for the past several years, because it complexes with cyclin D (a G1 cyclin). This complex is an important element in the p16INK4A-retinoblastoma (RB) gene pathway, which is commonly disrupted in cancer. Interestingly, the neural cdk5 is also expressed at high levels in certain cancer types (e.g., myeloma), potentially representing a therapeutic target in these diseases.21 Moreover, cdk5 may serve as a prognostic marker and help to identify patients most likely to respond to treatment (e.g., with bortezomib).22 In addition, inhibition of cdk1 and/or cdk5 may contribute to disruption of the unfolded protein response (UPR) secondary to endoplasmic reticulum (ER) stress inducers.23 Three transcription-regulatory cdks have been characterized. Cdk7 interacts with cyclin H and is responsible for phosphorylating cdks on threonine residues.24 Cdk8 interacts with cyclin C, Med12, and Med13, and forms a complex called “cdk8” subcomplex. Several substrates for cdk8, when complexed with the above-mentioned proteins, have been detected, including RNA polymerase II and histone H3.25 Cdk8 directly antagonizes the repression of β-catenin transcription by the transcription factor E2F1 in colorectal cancer.26 The suppression of β-catenin by E2F1 contributes to apoptosis. Therefore, overexpression of cdk8 (and RB) accounts for a reduced rate of apoptosis and increased cell growth.26 Cdk9 binds cyclin T and displays a tissue-specific expression pattern.27 That cdk9/cyclin T specifically interacts with the tat element of HIV-1 links this cyclin-dependent kinase directly to the replication pathway of HIV, and circumstantially to HIV-1–related malignancies (e.g., Kaposi sarcoma).28 Cdk1029 and cdk1130 define a novel class of cyclin-dependent kinases; both cdks interact with apoptosis-related factors30 or transcription factors such as ets.31 Cdk10 has two isoforms with different functions. A role at the G2/M transition has been suggested for the first isoform of cdk10, whereas the alternative splicing form interacts with the N-terminus of the Ets2 transcription factor. This interaction affects the G2/M transition in mice.32 Two known cyclin-dependent kinases, cdk12 and cdk13, interact with both forms of cyclin L (cyclin L1 and L2). This complex seems to be involved in alterative RNA-splicing,10,33 and thus is involved in the pre–messenger RNA processing machinery. All cyclins share an approximately 150-amino-acid region, called the cyclin box, which interacts with the cdks.34 The G1 cyclins (C, D, and E) and the mitotic cyclins (A and B)35 form distinct categories, although cyclin H, cyclins L1 and L2, and the type T cyclins (T1, T2a, and T2b) fall outside these two major groups.
Cyclin A binds and activates cdk2 mainly in S phase. However, microinjection of anti–cyclin A antibodies into cells causes cell cycle arrest just before S phase.11 This observation, together with the finding that overexpression of cyclin A leads to accelerated S-phase entry, suggests that cyclin A is involved in transformation.13 Cyclin A is able to compensate for loss of cyclin E function. Cyclin E is important for the duplication of centrosomes. In cyclin E-defective cells, cyclin A can take over the function of cyclin E in S phase, whereas cyclin A is important for centrosome amplification in G2-arrested cells, irrespective of whether cyclin E is present.36 The importance of cyclin A in cell division is underlined by other reports.37 In addition to its role at the G1/S boundary, cyclin A acts in late G2 phase, where it complexes with cdk1. Cyclin E, the other cyclin that interacts with cdk2, may control the progression from G1 to S phase, and the time point when cdk2 “switches” from cyclin E to cyclin A binding is right after prereplication complex assembly terminates while DNA replication initiates. Cells overexpressing cyclin E progress much faster through G1 into S phase, but the time required for DNA synthesis remains normal.38 On the other hand, a bifurcation in cdk2 activity determines whether cells immediately commit to the next cell cycle or enter a transient state of quiescence as they exit mitosis.39 Cyclin E levels are also regulated by environmental factors, including transforming growth factor-β (TGF-β) and irradiation. These effects are, in part, mediated by small proteins, the cyclin-dependent kinase inhibitors. Cyclin E accumulates at the G1/S boundary of the cell cycle, where it stimulates functions associated with entry into and progression through S phase.40 In normal cells, cyclin E levels are highly regulated so that peak cyclin E–cdk2 kinase activity occurs only for a short interval near the G1/S boundary.40 Cyclin E–cdk2 complexes become active during S phase and are then rapidly ubiquitinated after phosphorylation.41 The poor prognostic implications of overexpression of cyclin E has been observed in a variety of human malignancies,42 leading to a high cyclin E level throughout the cell cycle. The direct linkage between cyclin E overexpression and tumorigenesis is not completely understood. It has been suggested that the cyclin E–cdk2 complex phosphorylates and inactivates the RB protein43 or leads to genomic instability via generation of aneuploid cells.44 Cyclin E overexpression delays progression through early phases of mitosis and causes mitosis to be executed aberrantly, thus dysregulating mitotic progression.45
The B-type cyclins associate with cdk1 to form the classical mitotic cyclin–cdk complexes.46 Cyclin B is synthesized in S phase and accumulates, and in the midst of M phase is ubiquitinated and degraded, allowing the cell to exit from mitosis. The G2/M checkpoint is very often defective in malignant cells, leading to uncontrolled M-phase entry and aneuploidy. The cellular localization of the cdk1–cyclin B complexes is strictly cell-cycle–dependent. Although the complexes accumulate in the cytoplasm during G2 and S phase, they move to the nucleus in mitosis and bind to the mitotic spindle.47,48 The cyclin B family has different family members with distinct functions. At mitotic entry, cyclin B1–cdk1 promotes chromosome condensation, nuclear membrane dissolution, mitotic aster assembly, and Golgi breakdown, whereas cyclin B2–cdk1 can only induce Golgi disassembly.49 At prophase, cyclin B1 accumulates in the nucleus50 and then localizes to condensed chromatin, spindle microtubules, centrosomes, and chromatin during prometaphase.51 Distinct sequence elements are responsible for the localization of cyclin B1 to the chromatin, centrosomes, and kinetochores during mitosis.52
The three cyclin D molecules—D1, D2, and D3—function mainly in G1 phase, where they bind cdk4 and cdk6. These complexes phosphorylate RB, restraining its inhibitory effects on E2F and related transcription factors. Cyclin D1 is the major D cyclin in most cell types. All three cyclin D molecules act in late G1 phase, just before entry into S phase. Cyclin D1 also exhibits a variety of non–cell-cycle regulatory functions. For example, cyclin D1 regulates microRNA biogenesis by induction of Dicer, a central regulator of microRNA maturation.53 Many tumors have high cyclin D1 levels without amplification or mutation of the cyclin D1 structural gene. Instead, cyclin D levels may be regulated by a feedback loop dependent on RB. Alterations of the RB gene in cancer may secondarily cause upregulation of cyclin D transcription. As a result of its central role in cell-cycle control, the cyclin D–cdk4 complex is an important target for anticancer drugs. Mice lacking cyclin D1 are completely resistant to ErbB-2–driven breast cancer.54 ErbB-2–induced mammary tumor development is also prohibited by the inactivation of the cyclin D1 partner cdk4, underlining the role of this complex in human malignancies.55 As aberrations of the p16–cdk4–cyclin D-RB pathway are common in the majority of cancers, the development of selective cdk4 inhibitors (e.g., palbociclib) launched promising efforts to target tumors displaying either cyclin D1 overexpression (e.g., breast cancer, mantle cell lymphoma, multiple myeloma) or cdk4 amplification (e.g., liposarcoma).56 Moreover, cyclin D1–cdk4 is also involved in regulation of glucose metabolism in postmitotic cells, suggesting a novel cell-cycle–independent function of this complex.57 However, cdk6, a functional homologue of cdk4, may also play an important role in tumorigenesis under certain circumstances. For example, acute myelogenous leukemia (AML) cells carrying mixed-lineage leukemia (MLL) rearrangements (e.g., MLL-AF9, MLL-AF4, and MLL-AF6) specifically rely on cdk6, rather than cdk4, to proliferate,58 suggesting that cdk6 might represent a target in MLL-driven leukemia.59 Interestingly, SUMOylation stabilizes the cdk6 protein, which may contribute to progression of some tumors (e.g., glioblastoma).60 Notably, cyclin D–dependent cdk4/6 also phosphorylates a variety of substrates (e.g., RB1 and its relatives RBL1 and RBL2, SMAD2, SMAD3, FOXM1, MEP50, etc.), forming a central node in a complex signaling network that governs the overall transcriptional and biologic response of cells to activation or inhibition of the kinases.61
Cdk7 plays dual functions in regulation of cell cycle and gene transcription. In the former case, cdk7, together with its partner cyclin H, acts as a cdk-activating kinase, which fully activates various cell-cycle–regulatory cdks (e.g., cdk1, cdk2, cdk4, and cdk6) by phosphorylating their T loop in a context-specific manner.62 For example, whereas cdk7 is required to determine cyclin specificity and activation order of cdc1 and cdk2 during S and G2 phases, it is also required to maintain the activity of cdk4 as cells exiting quiescence and G1 progression through the restriction point.63 Cdk7, as a component of the general transcription factor TFIIH (cdk7/cyclin H/Mat1 complex), phosphorylates the carboxy-terminal domain (CTD, serines 5 and 7) of RNA polymerase II, which is responsible for transcription initiation and promoter clearance, a critical step in switching initiation to elongation during transcription.64 As transcription factors that co-opt the general transcriptional machinery to sustain the oncogenic state, pharmacologic inhibition of cdk7 may represent a novel approach to treat tumor types (e.g., T-cell acute lymphoblastic leukemia [T-ALL]) that are particularly dependent on transcription.65
Cdk9, as a catalytic subunit, partners with the regulatory subunit cyclin T, an 87-kDa cyclin C–related protein with three isoforms, to form a complex known as positive transcription elongation factor b (P-TEFb).66 The so-called cdk9-related pathway consists of two cdk9 isoforms (cdk9–42 and cdk9–55), cyclin T1, cyclin T2a, cyclin T2b, and cyclin K.27 Cdk9 and its binding partner cyclin T1 comprise the P-TEFb.67 P-TEFb hyperphosphorylates the CTD (primarily serine 2) of RNA polymerase II, essential for transcription elongation. P-TEFb also phosphorylates the negative transcription elongation factors (N-TEFs), including DRB-sensitivity inducing factor (DSIF) and the negative elongation factor (NELF), to release the transcription block (a pause immediately after transcription initiation) of both N-TEFs on hypophosphorylated forms of RNA polymerase II. In addition, cdk9 also plays a role in ribosomal RNA processing through activation of RNA polymerase II.68 In normal cells, the activity of P-TEFb is stringently maintained in a functional equilibrium to accommodate transcriptional demands for different biologic activities.69 As a rule in oncogenic transformation, upregulated antiapoptotic or prosurvival proteins in transformed cells must be sustained by constitutive RNA polymerase II activity that governs transcription elongation, in which cdk9 is the primary processivity factor.70 In other words, transformed cells are addicted to transcription because of the requirement for continuous production of antiapoptotic proteins, particularly those with short half-lives. Of note, abnormal activities in the cdk9-related pathway occur in many human malignancies.71 For example, high levels of cdk9/cyclin T1 expression are found in several types of hematologic malignancies, including B- and T-cell precursor-derived lymphomas, anaplastic large cell lymphoma, and follicular lymphomas, whereas strong nuclear staining for both proteins are observed in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. In this context, selective cdk9 inhibitors preferentially target malignant cells in preclinical hematologic tumor models, including leukemia and multiple myeloma.72,73 Mcl-1, the Bcl-2 family antiapoptotic protein with an estimated half-life of less than 3 hours,74 represents one of the most common downstream targets for cdk9 inhibition.75 Moreover, cdk9 inhibition disrupts the process of cytoprotective autophagy, for example, through downregulation of the adaptor protein SQSTM1/p62, resulting in an inefficient form of autophagy from cargo-loading failure, which, in turn, triggers apoptosis via upregulation of the BH3-only protein NBK/Bik.76 Moreover, cdk inhibitors also induce upregulation of other BH3-only proteins such as Bim and Noxa.77 Although it remains to be determined whether these events stem from inhibition of specific cdk(s), it is now clear that in addition to cell cycle-regulatory cdks, transcription-regulatory cdks, such as cdk9, represent another class of therapeutic targets. In addition, P-TEFb forms a complex with the HIV tat protein that binds the transactivation response element. The modification of RNA polymerase II by cdk9/cyclin T facilitates the efficient multiplication of the viral genome.78 Other binding partners of cdk9 include tumor necrosis factor receptor-associated factor 2,79 as well as inhibitory MAQ1 (or HEXIM1) and 7SK small nuclear RNA.80 Furthermore, cdk9 is expressed throughout the cell cycle81 and is also involved in viral (HIV, herpes) replication.27
Other members of the transcription-regulatory cdk subfamily include cdk8 and cdk12. Cdk8 is a subunit of the large Mediator complex (~1.2 MDa) composed of 25 to 30 proteins, which acts as a molecular bridge between DNA-binding transcription factors and RNA polymerase II. Cdk8 binds to cyclin C, MED12, and MED13 in the cyclin C–cdk8 module of the Mediator.82 The cdk8/cyclin C pair facilitates phosphorylation of both serine 2 and serine 5 at the CTD of RNA polymerase II.83 Cdk8 can perform both positive and negative functions in transcriptional regulation during different transcription stages (e.g., preinitiation and elongation), which provide a mechanism to respond to different promoter contexts (e.g., transcription factors or cdk8 module binding).82 Unlike cdk7 and cdk9, which govern global gene expression, cdk8 promotes only gene-specific transcription. Recently, cdk8 expression has been detected in 70 percent colorectal cancers and correlated with β-catenin activation, suggesting that cdk8 may act as a oncogene in certain types of cancer (e.g., colorectal and pancreatic cancer).84,85 Cdk12 and cdk13 have been identified as CTD kinases, both of which are unusually large proteins that contain a central kinase domain and share the same partner, cyclin K.86 Cdk12/cyclin K1 phosphorylates the CTD (preferably serine 2) of RNA polymerase II.87 Interestingly, cdk12/cyclin K only regulates expression of a small subset of genes, predominantly long genes with high exon numbers and DDR genes, including critical regulators of genomic stability, for example, BRCA1, ATR (ataxia-telangiectasia mutated [ATM] and Rad3 related), FANCI, and FANCD2.88
The cdk10 gene encodes two different cdk-like putative kinases; it is postulated that they exert their function at the G2/M transition.29 These two isoforms predominate in human tissues, except in brain and muscle, and the relative isoform levels do not vary during the cell cycle.29 Cdk10 interacts with the N-terminus of the Ets2 transcription factor, which contains the highly conserved pointed transactivation domain. The pointed domain is implicated in protein–protein interactions and Ets2 requires an intact pointed domain to bind Cdk10, which inhibits Ets2 transactivation in mammalian cells.31 This could be an important factor for the development of follicular lymphoma, because cdk10 is overexpressed in this cancer.89 In addition, cdk10 silencing increases Ets2-driven transcription of c-RAF, resulting in mitogen-activated protein kinase (MAPK) pathway activation and loss of tumor cell reliance upon estrogen signaling.90 Cdk10 promoters are frequently hypermethylated in malignant tumors, resulting in low expression levels of cdk10 and impaired cell-cycle regulation.90
Cdk11 is associated with cyclin L.91 It is part of the large family of p34(cdc2)-related kinases whose functions appear to be linked with cell-cycle progression, tumorigenesis, and apoptotic signaling. Cdk11 interacts with the p47 subunit of eukaryotic initiation factor 3 during apoptosis and is therefore directly involved in cell death mechanisms.92 Casein kinase 2 phosphorylates the cdk11 aminoterminal domain, suggesting that cdk11 participates in signaling pathways that include casein kinase 2 and that its function may help to coordinate the regulation of RNA transcription and processing events.91 So far two isoforms of cdk11 have been identified, a larger p110 and a smaller p46 isoform. During Fas- or tumor necrosis factor-α–induced apoptosis, the caspase-processed p46 isoform is generated from the larger p110 isoform and it promotes apoptosis when it is ectopically expressed in human cells. Cdk11 also stabilizes the microtubule assembly of cells93; cdk11 is therefore mandatory for the maintenance of sister chromatid cohesion94 and its disruption can contribute to the development of cancer.95
Many cyclin–cdk substrates have been identified by immunoprecipitation or two-hybrid assays, but only a few of them are thought to exert a direct function in cell-cycle control. The regulation of the cell cycle has been studied extensively during the last decade and a consensus paradigm of cell-cycle regulation has been suggested.50,96 According to this paradigm, the important switch of the cell cycle is the RB family of proteins (Fig. 16–2). In its hypophosphorylated state, RB binds to and inhibits a class of transcription factors, of which the best characterized is the E2F transcription factor. Hyperphosphorylation causes RB to detach from its binding site, permitting transcriptional activation of genes necessary for DNA synthesis and cell division. This phosphorylation of RB is regulated in a cell-cycle–dependent manner.97 A widely accepted model suggests that RB is phosphorylated by different regulators such as cyclin E/cdk2 at the so-called “R” point, a time point during G1 when cell cycle progression becomes independent of exogenous stimuli.98 Interference with RB function impairs G1 checkpoint regulation and fosters unrestrained cell growth, a nearly universal characteristic of malignancy. RB controls the activity of several other cell-cycle regulatory elements such as Skp2.99 The Skp2 regulation follows an autocrine loop where Skp2 triggers degradation of the cdk inhibitor p27 kip1, followed by cyclin E/cdk2 activation, consecutive cdk2-induced RB-phosphorylation, and further E2F-dependent Skp2 expression.100 Causes of reduced RB activity include changes in the structural gene, the sequestration and inactivation of the protein by viral oncogene products, and hyperphosphorylation of RB as a result of increased cdk4 and cyclin D activity or deletion of the gene for the p16INK4A inhibitor of cdk4. Deletions, mutations, and translocations of RB are common in various malignancies, while homozygous deletions of the p16INK4A gene are even more frequent. Many different transforming viruses (papillomavirus, simian virus 40) produce proteins that interact with RB. Both cyclin D1–cdk4 and cyclin D1(D2, D3)–cdk6 complexes are able to phosphorylate RB.101,102 The time point of RB phosphorylation correlates strongly with the appearance of the cyclin D1–cdk4 complex.103 The link between RB and cyclin D is supported by the observation that loss of RB function leads to a decrease in the cellular cyclin D level.104 However, cyclin D is not the only cyclin that is involved in the RB regulatory pathway.99,102 Ectopic expression of both cyclin A and cyclin E restores RB hyperphosphorylation and causes cell-cycle arrest in cancer cell lines. Perhaps the cdk2–cyclin A complex contributes to additional phosphorylation of RB, whereas the cdk2–cyclin E complex prolongs the phosphorylation time.105
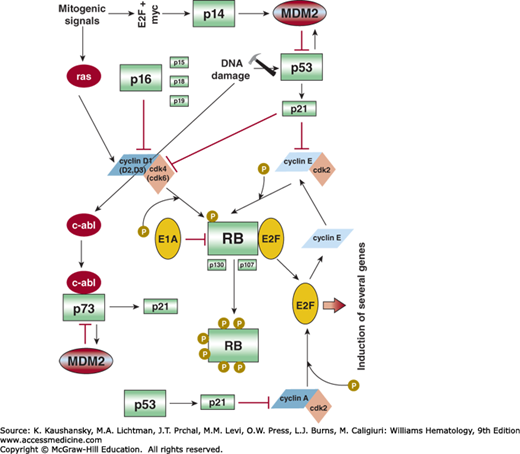
The key regulatory element for the G1-to-S transition is the RB–E2F complex. After RB is phosphorylated by cdk4 and/or cdk6 complexes during G1 phase and cdk2 at G1/S interphase, E2F proteins are released and promote the transcription of genes essential for the transition to S phase.99,106 As mentioned above, the p16INK4A/cyclin D1/cdk4/RB/E2F cascade is probably one of the most important cascades in cell-cycle control, and is frequently affected in human cancer. For example, this pathway is defective in nearly 100 percent of AML cell lines and most of the primary AML samples, although the exact mechanism of inactivation is not always clear. Two RB-related pocket proteins, p107 and p130, also form complexes with the transcription factor E2F,107 bind to the region of the adenovirus E1A protein required for transformation, and are able to induce G1 arrest when they are overexpressed in human malignant cell lines.108,109 Unlike RB, the p107 and p130 proteins contain a so-called spacer region that interacts with cdk2/cyclin A and cdk2/cyclin E,110 although it seems to be unlikely that these two complexes regulate the activity of p107 and p130.105 Instead, p107 may bind and inactivate the cyclin A and cyclin E complexes. Thus, p107 may regulate the cell cycle by several different mechanisms. Because both p107 and p130 are regulated through phosphorylation, efficient cell-cycle entry is accompanied by phosphorylation of all the RB-related proteins.111
In addition to its cell-cycle regulatory properties, RB also influences hematopoietic differentiation.112 RB interacts with the transcription factor PU.1, which blocks erythroid differentiation in the proerythroblast stage when ectopically overexpressed in marrow cells,113,114 and represses GATA-1 activity.115 An important event in this differentiation process is the interaction between hematopoietic stem cells and the microenvironment of the marrow. In addition, hypophosphorylated RB promotes monocytic over neutrophilic differentiation in bipotent progenitor cells, an event that is switched to neutrophilic differentiation if RB expression is inhibited. This finding points to an important property of RB independent of cell-cycle control.116
Besides regulation by phosphorylation, specific protein inhibitors of cdk enzymatic activity have been identified.117 The cyclin-dependent kinase inhibitors (CDKIs) cause cells to arrest in G1 phase, followed by differentiation and/or senescence. The first CDKI identified was p21cip1.118 It binds to several cyclin/cdk complexes, including cyclin A/cdk2, cyclin D/cdk4, and cyclin E/cdk2 (see Fig. 16–2).97,119 Several different cell-cycle regulatory pathways are affected by p21cip1. In addition, a basal p21cip1–cdk2 axis determines quiescent and cycling cell states and thus controls population heterogeneity in both normal cells and tumors, which can make anticancer treatment selectivity challenging.120 The molecule has a p53 binding site in its promoter, and an increase in p53 levels results in transcriptional activation of p21cip1, slowing cell-cycle progression. In addition to this p53-dependent pathway, p21cip1 is also regulated in a p53-independent manner. For example, histone deacetylase inhibitors (HDACIs) are able to induce p21cip1 expression in p53null leukemia cells through an alternative nuclear factor (NF)-κB–dependent mechanism, which may limit the antileukemia activity of these agents.121,122 Several binding partners of p21cip1, including Pim-1, have been identified. Pim-1 associates with and phosphorylates p21cip1 in vivo, which influences the subcellular localization of p21cip1.123 p21cip1 is phosphorylated by Pim-1 at two distinct sites, Thr145 and Ser146; phosphorylation on Thr145 results in a nuclear localization of p21cip1 and a disruption of the cell cycle, while phosphorylation on Ser146 leads to a cytoplasmic localization of p21cip1,124 suggesting that overexpression of Pim-1 in certain tumors plays a key role in tumorigenesis.125 Like RB, expression and function of p21cip1 is also affected by several different mechanisms, including mutation and histone deacetylation (see “The Role of Histone Deacetylases in Cell-Cycle Regulation” below). Other members of the p21cip1 family of CDKIs include p27kip1 and p57kip2.101,126 As a cdk inhibitor, p27kip1 has tumor-suppressor activity. Besides cdks, p27kip1 regulates additional cellular processes, including cell motility, some of which seem to mediate the oncogenic activities of p27kip1. For example, the constellation of high p27kip1 and low Myc expression is characteristic of chronic lymphocytic leukemia (CLL) cells.127 These activities of p27kip1 are regulated through multiple phosphorylation sites. The multiple functions of p27kip1 are dependent on a number of different conditions, and dictate whether the protein displays anti- or protumorigenic properties.128 High-level expression of p27kip1 leads to a cell-cycle block in G1 phase after treatment of cells with TGF-β. One major difference between p21cip1 and p27kip1 is that the former binds predominantly to cdk2 whereas the latter binds cdk4.
The cellular levels of a number of cell-cycle regulators, including p21cip1 and p27kip1, are regulated by ubiquitination and subsequent proteolysis. Polyubiquitinated proteins are degraded by the 26S proteasome complex. There are two major ubiquitination systems in the cell; they are designated SCF and APC.108,110 SCF is named for three of its core components, Skp, Cullin, and an F-box–containing protein. Important examples of SCF substrates are Cln1, Sic1, Wee1, Cdc6/Cdc18, E2F, cyclin D1, cyclin E, p21cip1, p27kip1, and p57kip2.129
A second group of CDKIs belongs to the inhibitor of the kinase 4 (INK4) family and includes p15INK4B, p16INK4A, p18INK4C, and p19INK4D.104,107,130,131 They all bind and inhibit the cyclin D1–cdk4 and/or cyclin D1–cdk6 complex, which regulate cell-cycle progression via RB.104,130 TGF-β is also a potent inducer of p15INK4B,104 one of the mechanisms by which the cytokine regulates the proliferation of hematopoietic cells (Chap. 16). p16INK4A is probably the most important CDKI, because the gene is inactivated by several mechanisms (deletion, mutation, hypermethylation) in many different human cancers.132 Surprisingly, p16INK4A and p14ARF are overexpressed in some cases of human hematologic malignancies.133 This overexpression is probably a result of defects downstream of p16INK4A, particularly caused by mutations in the RB gene.134 In hematologic malignancies, the highest frequencies of p14ARF, p15INK4B, or p16INK4A inactivations are found in T-ALL,135 secondary high-grade lymphomas, and mantle cell lymphoma (MCL).136,137 The potency of p14ARF and p16INK4A in terms of tumorigenicity becomes obvious because the reexpression of both genes by either retroviral transfection or demethylation of the promoter regions results in a complete reversion of the malignant phenotype.138,139 p14ARF has multiple tumor-suppressor functions, some of which are mediated by signaling to p53. On the other hand, it has been shown that p14ARF is able to drive tumor progression in a p53-independent fashion, especially in myc-driven lymphomas.140
PHARMACOLOGIC INHIBITION OF CYCLIN-DEPENDENT KINASES
Perturbations of the cell cycle, for example, overexpression of cyclins or underexpression of endogenous CDKIs, are nearly universal in human malignancies, and cdks, as critical regulators of cell-cycle progression and RNA transcription, represent attractive targets for anticancer drug development, as their inhibition can lead to both cell-cycle arrest and apoptosis.141,142 Additionally, pharmacologic inhibition of transcriptional cdks, for example, cdk9, affects proteins with short half-lives, for example, the antiapoptotic proteins Mcl-1 and XIAP (X-linked inhibitor of apoptosis), cell-cycle regulators, p53, and NF-κB–responsive gene products.142 Hematologic malignancies may be particularly susceptible to cdk inhibition and apoptosis induction. A number of pharmacologic CDKIs have been developed and subjected to various phases of preclinical and clinical testing in hematologic malignancies (for a review, see Ref. 143). The selective cdk4/6 inhibitor palbociclib has been approved by the FDA as a potential treatment for hormone-responsive metastatic breast cancer. In MCL, a disease characterized by cyclin D1 overexpression, this agent has demonstrated encouraging clinical activity.144 Whether selective or broad-spectrum inhibition is the superior therapeutic strategy continues to be debated,143 and may be tumor type-specific. The “pan-CDKI” flavopiridol (alvocidib), the first CDKI to enter the clinic, has shown promising activity in patients with genetically high-risk chronic lymphocytic leukemia (CLL), particularly when administered by a pharmacologically derived “hybrid” schedule,145,146 but recent developmental efforts for this agent have focused on AML, where it was recently granted “orphan drug” status.147 Flavopiridol induces apoptosis in primary leukemic blasts and recruits surviving leukemic cells into a proliferative state, thereby priming such cells for killing by S-phase-active cytotoxic agents such as cytarabine.148,149 These observations led to the design of “timed sequential therapy” regimens, for example, FLAM (flavopiridol, cytarabine, mitoxantrone), which exhibited promising clinical activity in AML patients with nonfavorable cytogenetics.150 Interestingly, both “bolus” and “hybrid” schedules of administration of flavopiridol produce comparably encouraging results in this setting.151 Other rational combinations involving pan-CDKIs such as flavopiridol or roscovitine involve those with proteasome inhibitors (PIs),152,153 HDACIs154 or BH3-mimetics.75 The first strategy has been explored in phase I trials in patients with recurrent or refractory indolent B-cell neoplasms,155,156 and a phase I trial of bortezomib and dexamethasone plus the pan-CDKI dinaciclib in patients with relapsed myeloma is ongoing as of this writing (NCT01711528). Synergism in preclinical studies in leukemia between pan-CDKIs and HDACIs is based predominantly on reciprocal effects on the cytoprotective NF-κB pathway and the endogenous cdk inhibitor p21WAF1/CIP1, besides Mcl-1/XIAP downregulation.154 Some of these phenomena have been recapitulated in patients with AML.157
CELL-CYCLE CHECKPOINTS
The cell cycle progresses in an orderly fashion and is monitored by safety mechanisms known as cell-cycle checkpoints, which, upon activation, function to halt cell division.158 When DNA damage occurs, distinct, albeit overlapping and cooperating, checkpoint pathways are activated, which block S-phase entry (the G1/S-phase checkpoint), delay S-phase progression (the intra–S- or S-phase checkpoint), or prevent mitotic entry (the G2/M-phase checkpoint). These events direct phase-specific DNA repair mechanisms through repair-specific gene transcription. If repair fails, checkpoints trigger apoptosis.159 Checkpoints are thus important quality control measures that ensure the proper sequence of cell-cycle events and allow cells to respond to DNA damage.160
The G1/S checkpoint is the first defense against genomic stress in cycling cells. In response to DNA damage, the G1/S checkpoint prevents cells from entering the S-phase by inhibiting the initiation of DNA replication. At this checkpoint, the checkpoint kinase Chk2 is activated by the proximal transducer ATM to phosphorylate (and thereby inhibit) the cdc25A phosphatase, thus preventing activation of cyclin E(A)/cdk2 and temporarily halting the cell cycle. G1 arrest is sustained by ATM/Chk2-mediated phosphorylation of murine double minute protein 2 (MDM2) and p53, resulting in p53 stabilization and accumulation. p53 transcriptionally activates the endogenous cdk inhibitor p21, which, in turn, inhibits cyclin E(A)/cdk2 and preserves the association of Rb with E2F.160
The intra-S or S-phase checkpoint is activated in response to structural DNA damage as well as stalled replication forks. Upon activation of this checkpoint, ATR/Chk1 and ATM/Chk2 phosphorylate cdc25A, resulting in enhanced proteolysis of the phosphatase and inhibition of its function through 14-3-3 σ binding. Cyclin E(A)/cdk2 is thereby inhibited, and progression through the S-phase halted.158
The G2/M checkpoint prevents mitotic entry of cells that have either incurred DNA damage during G2, or that have escaped the G1/S and intra-S checkpoints despite earlier genomic insults. The key downstream target of the G2/M checkpoint is the promitotic cyclin B/cdk1 (cdc2) complex. During interphase, this complex is inactivated through phosphorylation by Myt1 and Wee1.158 Chk1 may phosphorylate Wee1, mediating binding of Wee1 to 14-3-3 proteins, which, in turn, may stimulate the kinase activity of Wee1 against cdk1 (cdc2). Thus, both Chk1 and 14-3-3 proteins may act together as positive regulators of Wee1.161 Activation of cyclin B/cdk1 (cdc2) requires dephosphorylation by the cdc25 phosphatases (A, B, and C). Notably, phosphorylation/inactivation of cdk1 (cdc2) involves two inhibitory sites, for example, Tyr15 and Thr14, and dephosphorylation of both sites is necessary for full cdk1 (cdc2) activation. Initiation of the G2/M checkpoint is mediated by ATR/Chk1, which phosphorylates (and thereby inhibits) cdc25A, B, and C, whereas maintenance of this checkpoint requires p53 and its downstream effectors p21, 14-3-3 σ, and growth arrest and DNA damage (GADD) 45.158
Checkpoint dysfunction is common in human cancers and is considered a pathologic hallmark of neoplastic transformation.162 Conversely, agents used for cancer treatment, such as cytotoxic chemotherapy and ionizing radiation, activate cell-cycle checkpoints.158 Cancer cells are particularly dependent upon the S- and G2/M-phase checkpoints for repair of DNA damage because of preexisting defects in G1/S checkpoint mechanisms, such as p53 and RB mutations. Because the S-phase checkpoint facilitates slowing, rather than arrest, of the cell cycle, a cancer cell harboring DNA damage may progress through the S-phase checkpoint, only to halt at the G2/M checkpoint. The latter is, therefore, a key guardian of the cancer cell genome, and its abrogation can lead to enhanced tumor cell death while sparing normal cells, which maintain an intact G1/S-phase checkpoint. G2/M checkpoint abrogation prevents cancer cells from repairing DNA damage, forcing them into a premature and lethal mitosis (“mitotic catastrophe”).160
Based upon these concepts, synergism between DNA-damaging agents (cytotoxic chemotherapy) and G2/M checkpoint abrogators have been examined both preclinically and in clinical studies. In human AML cell lines, the Chk1 inhibitor MK-8776 (SCH900776) markedly increased cytarabine-induced apoptosis, with negligible impact on normal myeloid progenitors,163 leading to a phase I trial of the combination in patients with relapsed and refractory acute leukemias.164 Chk1 inhibitors increase HDACI lethality in human leukemia cells.165 This may reflect the ability of HDACIs to induce DNA damage in AML cells,166 inhibit both homologous recombination and nonhomologous end-joining mechanisms of DNA repair,167 and downregulate/inactivate Chk1.168 However, clinical development of MK-8776 has been halted. Based upon the ability of Hsp90 inhibitors to downregulate Chk1, a phase I study of the combination of cytarabine and the Hsp90 inhibitor tanespimycin was conducted in adults with recurrent or refractory acute leukemia; however, the combination exhibited limited clinical activity.169
Wee1 is a validated therapeutic target in AML.170 The combination of cytarabine and the Wee1 inhibitor AZD1775 synergistically induces apoptosis in myeloid cell lines.171 Similarly, genome-wide short hairpin RNA screens strongly implicate cell-cycle checkpoint proteins, particularly Wee1, as critical mediators of AML cell survival after cytarabine exposure.172 In AML cell lines, synergistic inhibition of proliferation by the combination of AZD1775 and Ara-C occurred regardless of p53 functionality.173 Another combinatorial strategy in AML involves coadministration of AZD1775 with HDACIs.174 Besides inducing DNA damage and inhibiting DNA repair, HDACIs downregulate several proteins critical to checkpoint function, such as ATR, Chk1, and Wee1,168,175,176,177 and Wee1 inhibition promotes premature mitotic entry of cells bearing unrepaired DNA damage.178 Efforts to extend these pre-clinical findings to the clinic are underway.
ONCOGENES
Table 16–2 lists the common genomic aberrations seen in hematologic malignancies. Tables 16–3 and 16–4 list common somatic mutations encountered in the major myeloid and lymphoid malignancies, respectively.
| Chromosomal abnormality | Genes/loci affected or fusion gene where applicable | Functional consequence, if known | Approximate incidence (in newly diagnosed patients) | Prognostic/therapeutic implications, if any |
|---|---|---|---|---|
| ACUTE MYELOID LEUKEMIA | ||||
| t(8;21)(q22;q22) – diagnostic of AML regardless of blast count | RUNX1-RUNX1T1 (AML1-ETO) | Fusion protein alters transcriptional regulation of normal RUNX1 target genes and activates new target genes to prevent apoptosis and/or differentiation | 7% in adults; most frequent abnormality in children with AML | Favorable prognosis in adults(not children) unless c-kit mutated; respond particularly well to high dose cytarabine-based regimens; generally not allografted in CR1 |
| inv(16)(p13.1q22) or t(16;16) (p13.1;q22) – diagnostic of AML regardless of blast count | CBFB-MYH11 | Fusion protein disrupts function of RUNX1/CBFB transcription factor→repression of transcription; association with M4 FAB subtype and abnormal bone marrow eosinophils | 5% in adults | Favorable prognosis unless c-kit mutated; respond particularly well to high dose cytarabine-based regimens; generally not allografted in CR1 |
| t(15;17)(q24.1;q21.1) – diagnostic of AML (APL) regardless of blast count | PML-RARA | Fusion protein causes transcriptional repression, preventing differentiation of promyelocytes | 13% (APL, most common form of AML related to therapy with bimolane for psoriasis) | Differentiation block overcome by pharmacologic doses of ATRA; high cure rates; low/intermediate risk patients can be cured without chemotherapy |
| 11q23 rearrangements | KMT2A (MLL) and various partner genes, e.g., AF9 (most common partner) or partial tandem duplication | Complex effects (CDKs important in leukemogenesis); can also result in lymphoid phenotype (e.g., KMT2A-AF4 fusion with t(4;11)(q21;q23) | 6% in young adults and up to 12% in children; often seen in AML following therapy with DNA topoisomerase II inhibitors | Poor prognosis unless t(9;11)(p22;q23) leading to KMT2A-AF9 fusion (intermediate prognosis); MLL translocations predict improved outcome with high dose daunorubicin |
| inv(3)(q21q26.2) or t(3;3)(q21;q26.2) or ins(5;3)(q14;q21q26.2) | MECOM (EV1) | MECOM (EV1) activation can promote or repress transcription depending on binding partners through interaction with transcriptional and epigenetic regulators | 1% (3q abnormalities as a whole in 4.4%) | Adverse prognosis; association with thrombocytosis and increased, often abnormal, bone marrow megakaryocytes |
| t(6;9)(p23;q34) | DEK-NUP214 (DEK-CAN) | Fusion protein is a nucleoporin that acts as a transcription factor and also alters nuclear transport | 1% | Poor prognosis; high incidence of FLT3-ITD; association with basophilia, dysplasia and pancytopenia |
| Trisomy 8 | 13% | Intermediate prognosis | ||
| Loss of chromosome 7 or del7q | CUX1, EZH2 | CUX1 encodes a transcription factor and EZH2 a histone methyltransferase; both act as tumor suppressors | -7 seen in 9% | Poor prognosis; strong associations with older age, secondary AML, complex karyotype and prior therapy with alkylating agents or radiation |
| Loss of chromosome 5 or del5q | RPS14, miR-145/146a, EGR1, NPM1, APC, CTNNA1 | Unclear; cooperative loss of multiple genes on 5q likely required for pathogenesis | -5 seen in 6% | Poor prognosis; strong associations with older age, secondary AML, complex karyotype and with prior therapy with alkylating agents or radiation |
| del17p | TP53 | Loss of tumor suppressor “guardian of genome”, failure of cell cycle checkpoint mechanisms, disruption of DDR | TP53 mutations seen in only 9% of older patients with de novo AML, but much more common in secondary and t-AML | Very poor prognosis, high risk of treatment failure not overcome even by allogeneic HSCT; strong association with complex karyotype |
| Complex karyotype (≥3 acquired, unrelated abnormalities in the absence of t(8;21), inv(16)/t(16;16) or t(15;17)) | Multiple | 10-12%, increases with age; much more common in secondary or t-AML | Very poor prognosis; 5q, 17p and 7q abnormalities most common; TP53 alterations the most important prognostic factor | |
| Monosomal karyotype (≥2 monosomies, or a single monosomy in the presence of structural abnormalities) | Multiple | 13%, increases with age | Extremely poor prognosis not overcome even by allogeneic HSCT | |
| CHRONIC MYELOID LEUKEMIA | ||||
| t(9;22)(q34;q11) | BCR-ABL1 (vast majority of cases characterized by the p210 “major” fusion protein) | Fusion protein (constitutively active tyrosine kinase) drives all aspects of pathogenesis in chronic phase; other pathways also important in advanced phases | All cases (atypical CML, Bcr-Abl negative, is a separate entity) | High degree of “oncogene addiction” in chronic phase allows successful targeting of the Bcr-Abl kinase by small molecule inhibitors; outcomes dismal in blastic phase with loss of addiction to Bcr-Abl signaling |
| MYELODYSPLASTIC SYNDROMES | ||||
| del5q33.1 or del5q31 | RPS14, miR-145/146a, EGR1, APC, CTNNA1 | In 5q- syndrome, haploinsufficiency of RPS14 leads to impaired maturation of 40S ribosomal units, causing premature TP53-dependent apoptosis of erythroid precursors | 15% overall | Most common chromosomal abnormality in MDS; del5q31 more common with higher risk or t-MDS; del5q33.1 seen in the indolent 5q- syndrome responsive to lenalidomide; del5q considered “good” in IPSS-R |
| del7q or loss of chromosome 7 | EZH2? | Unclear loss of which genes on 7q pathogeneic, since vast majority of patients with EZH2 mutations do not have del7q or -7 | 10% in de novo MDS; up to 50% in t-MDS | del7q considered “intermediate” in IPSS-R; -7 considered “poor” |
| Trisomy 8 | c-MYC, others? | Higher expression of anti-apoptotic genes, c-MYC overexpression? | <10% | Considered “intermediate” in IPSS-R |
| del20q | Unknown | Unknown | <5% | Considered “good” in IPSS-R |
| Loss of Y chromosome | Multiple | Not felt to be pathogenic in MDS | Common normal finding in men | Considered “very good” in IPSS-R |
| Complex karyotype (≥3 abnormalities) | Multiple | 18% | Considered “very poor” in IPSS-R if >3 abnormalities; “poor” if only 3 | |
| t(5;12)(q33;p13) | ETV6-PDGFRB (TEL-PDGFRB) | Fuses ETV6 transcription factor gene with PDGFR beta gene | Rare | Seen in the context of MDS/MPN; responds to imatinib |
| B-CELL ACUTE LYMPHOBLASTIC LEUKEMIA | ||||
| t(9;22)(q34;q11) | BCR-ABL1 (most cases characterized by the p190 “minor” fusion protein | Fusion protein leads to constitutively active tyrosine kinase; often associated with IKZF1 splicing abnormalities | 25-30% in adults, 2-5% in children | Traditionally “high risk”, with allogeneic HSCT in CR1 commonly recommended; cure rates improved in “TKI era” |
t(8;14)(q24.2;q32) or t(2;8)(p12;q24.2) or t(8;22)(q24.2;q11.2) | c-MYC, immunoglobulin heavy and light chain genes | Constitutive expression of oncogenic c-MYC | 1% (Burkitt’s lymphoma/leukemia – FAB L3 morphology; mature B-cell phenotype) | Poor prognosis; high incidence of CNS involvement and TLS; treated with short, intensive regimens |
| t(12;21)(p12;q22) | ETV6-RUNX1 (TEL-AML1) | Fusion protein inhibits transactivation of gene expression by normal RUNX1 | 15-25% in children, 3-4% in adults | Favorable prognosis |
| Intrachromosomal amplification of chromosome 21 (secondary chromosomal abnormalities common) | RUNX1 (AML1) | 3-5% of older children | Poor prognosis with standard risk regimens, overcome by intensive therapy (high risk regimens) | |
| 11q23 abnormalities | KMT2A (MLL), most commonly KMT2A-AF4(AFF1) caused by t(4;11)(q21;q23); 2nd most common is t(11;19)(q23;p13.3) | KMT2A encodes a histone methyltransferase that regulates transcription of genes such as IKZF1 and HOX genes | 5-7% overall; 60-80% in infants | Poor prognosis; frequent co-expression of myeloid antigens; about half the patients with t(11;19)(q23;p13.3) have AML, mostly FAB M5a subtype |
| t(1;19)(q23;p13.3) or der(19)t(1;19)(q23;p13.3) and its variant t(17;19)(q21;p13.3) | PBX1-TCF3 (PBX1-E2A) or HLF-E2A | Activation of gene transcription by fusion protein via unclear mechanisms | 30% of childhood precursor B-cell ALL (5% of all pediatric ALL) | Studies with conflicting results; probably favorable prognosis |
| del9p | PAX5, JAK2 (in Down syndrome associated ALL) | 5-10% | Poor prognosis | |
| Hyperdiploidy | Multiple | 40% | Favorable prognosis in patients with >50 chromosomes | |
| Hypodiploidy | Multiple | 5-6% | Poor prognosis | |
| T-CELL ACUTE LYMPHOBLASTIC LEUKEMIA | ||||
| Translocations involving 7p14 (TCRγ) or 7q34(TCRβ) or 14q11.2 (TCRα/δ) | TCR genes; multiple partners – usually cell cycle inhibitors or transcription factors | 35% | ||
| Cryptic interstitial deletion at 1p32 | SIL-TAL1 | Fusion gene functions as transcription factor | 9-30% of childhood T-ALL; frequency decreases in adults | |
| 11q23 abnormalities | KMT2A (MLL) with various partners | 8% | ||
| t(10;11)(p13;q14) | CALM-AF10 | 10% | ||
| t(9;9)(q34;q34) | NUP214-ABL1 | Fusion of nuclear pore complex component with intracellular tyrosine kinase as a result of episomal amplification of in-frame fusion | Up to 6% | Responsive to imatinib? |
| del9p21 | p16 (INK4/ARF) | Loss of endogenous CDK inhibitor function | 65% in children; 15% in adults | |
| del6q | 20-30% | |||
| CHRONIC LYMPHOCYTIC LEUKEMIA | ||||
| del13q | MIR15A, MIR16-1, RB1, associated with MYD88 mutations | Loss of tumor suppressor function of Rb; loss of miR15a and miR16-1 derepresses Bcl-2 but also increases TP53 mRNA; MYD88 is a critical adaptor molecule of the IL-1 TLR pathway that activates NF-κB and augments BTK signaling | 55% | Favorable prognosis when sole cytogenetic abnormality; MYD88 mutations associated with mutated IGHV; BTK (BCR pathway) inhibition highly effective in CLL |
| del11q | ATM; associated with SF3B1 mutations | ATM is a proximal transducer of the DDR network; SF3B1 functions in mRNA splicing | 18% | Poor prognosis but overcome by FCR; the most common type of karyotypic evolution over time; associated with bulky lymphadenopathy |
| Trisomy 12q | Associated with NOTCH1 and FBXW7 mutations | 16% | Unclear – conflicting study results; NOTCH1 mutations confer unfavorable prognosis and are associated with unmutated IGHV | |
| del17p | TP53 | Loss of tumor suppressor “guardian of genome”, failure of cell cycle checkpoint mechanisms, disruption of DDR | 7% | Poor prognosis not overcome by cytotoxic chemotherapy; alemtuzumab and BCR pathway inhibitors effective, as well as allogeneic HSCT |
| del6q | 7% | |||
| T-CELL PROLYMPHOCYTIC LEUKEMIA | ||||
t(14;14)(q11;q32) or inv(14)(q11;q32) or t(X;14)(q28;q11) | TCR-TCL1 MTCP1-TCR | Overexpression of the TCL-1 oncogene or its homolog MTCP1 | 90% | |
| Chromosome 8 abnormalities | idic(8p11), t(8;8), trisomy 8q | Up-regulation of oncogenic c-MYC | 70-80% | |
| MULTIPLE MYELOMA | ||||
| del13q14 or monosomy 13 or hypodiploidy | Multiple | 48% | Intermediate risk with modern therapies only if detected on karyotyping, or by FISH in presence of other abnormalities (otherwise standard risk) | |
| del17p13 | TP53 | Loss of tumor suppressor “guardian of genome”, failure of cell cycle checkpoint mechanisms, disruption of DDR | 11% | High risk |
| Gain of 1q21 | CKS1B | CKS1B favors cell cycle progression by promoting degradation of the endogenous CDK inhibitor p27 with release of CDKs and mitotic entry | 30-43% | Controversial; conflicting study results |
| del1p | FAM46C, CDKN2C | Loss of endogenous CDK inhibitor function leads to excessive proliferation | 30% | High risk |
| t(11;14)(q13;q32) | CCND1-IgH | Overexpression of cyclin D1 | 21% (10-31%) | Standard risk; very high prevalence in non-secretory cases; associated with lower levels of monoclonal proteins, CD20 expression, lambda light chains and lymphoplasmacytic morphology |
| t(6;14)(p25;q32) | CCND3-IgH | Overexpression of cyclin D3 | Standard risk | |
| t(8;14)(q24;q32) | c-MYC-IgH | c-MYC overexpression | Standard risk | |
| t(4;14)(p16.3;q32.3) | MMSET-IgH | MMSET is a histone methyltransferase and its deregulation/overexpression is key to pathogenesis; FGFR3 also often overexpressed | 14% | Intermediate to high risk; adverse prognosis overcome by bortezomib/HSCT |
| t(14;16)(q32.3;q23) | IgH-c-MAF | MAF encodes a transcription factor that can activate or repress transcription depending on binding site/partner | High risk | |
| t(14;20)(q32;q11) | IgH-MAFB | MAF encodes a transcription factor that can activate or repress transcription depending on binding site/partner | High risk | |
| Hyperdiploidy (trisomies of odd numbered chromosomes other than 1, 13, 21) | Multiple | 39% | Standard risk; in the presence of concurrent trisomies, “high risk” cytogenetics become standard risk | |
| NON-HODGKIN’S LYMPHOMA (selected abnormalities) | ||||
| Chromosomal abnormality | Fusion gene created, where applicable | Pathogenetic mechanism | Disease association | Therapeutic relevance |
| t(2;5)(p23;q35) and variants | ALK-NPM and others | Constitutively active tyrosine kinase triggers malignant transformation and activates anti-apoptotic pathways | ALK+ anaplastic large cell lymphoma | ALK targeting with small molecule inhibitors, e.g., crizotinib |
| t(11;14)(q13;q32) | CCND1-IgH | Overexpression of cyclin D1 drives cellular proliferation | Mantle cell lymphoma (almost all cases) | Efficacy of CDK4/6 inhibitors, e.g., palbociclib |
| t(14;18)(q32;q21) | IgH-BCL2 | Constitutive expression of anti-apoptotic Bcl-2 promotes cellular survival | Follicular lymphoma (80%), diffuse large B-cell lymphoma (30%) | Selective targeting of Bcl-2 with BH3-mimetics, e.g., venetoclax |
t(11;18)(q21;q21) t(14;18)(q32;q21) t(1;14)(p22;q32) t(3;14)(p13;q32) | API2 (IAP2)-MALT1 IgH-MALT1 BCL10-IgH FOXP1-IgH | Overexpression of BCL10 results in NF-κB activation through BCL10/MALT1 signaling complex; FOXP1 is a transcription factor of unknown function | MALT lymphoma (extranodal marginal zone lymphoma) | Nuclear expression of BCL10 or NF-κB in gastric MALT lymphoma associated with resistance to antibiotic therapy |
| 9p24 amplification | Contains genes that encode PD-L1, PD-L2, JAK2 | PDL-1 commonly overexpressed; MHC class II transactivator rearrangements (38% of PMBL) lead to PD-1 overexpression also | Primary mediastinal B-cell lymphoma | PD-1 immune checkpoint pathway a potential target |
| 3q27 abnormalities (mutations, rearrangements) | Locus for BCL6 transcriptional repressor | Bcl-6 overexpression results in down-regulation of many target proteins, including p53 tumor suppressor | Diffuse large B-cell lymphoma (almost all cases) | New inhibitors that disrupt Bcl-6 function being developed |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


