FIGURE 6.1 Windows of cyclin-dependent kinase (CDK) function in the cell cycle. D-type cyclins (cyclins D1, D2, and D3) activate CDK4 and CDK6 for functions extending from middle G1 to the G1/S-phase transition. E-type cyclins (cyclins E1 and E2) activate CDK2 for functions at the G1/S-phase boundary, probably extending into early S phase. Cyclin A activates CDK2 for functions extending from the G1/S-phase boundary and extending into G2. Cyclin A is known to interact with CDK1 as well; however, no specific function for this complex has been identified. Finally, cyclin B activates CDK1 at the G2/M-phase boundary with activity that lasts until cyclin B is degraded during anaphase.
Unlike D-type cyclins, E-type cyclins (E1 and E2) are expressed with high cell-cycle periodicity, accumulating in late G1 and declining during S phase. E-type cyclins activate CDK2, and the fact that premature expression of cyclin E1 leads to accelerated entry into S phase10,11 has suggested that the target(s) must be proteins responsible for initiation of DNA replication. However, the essentiality of cyclin E–CDK2 in this context has been put into question by the demonstration that cells from cyclin E1/E2 nullizygous mouse embryos can cycle with reasonably normal kinetics and can certainly initiate DNA replication.12 The most likely explanation for the dispensability of E-type cyclins for S phase functions is redundancy with cyclin A, which also activates CDK2.
Cyclin A accumulates initially at the G1/S-phase boundary and persists until prometaphase of mitosis. It has been best characterized as an activator of CDK2; however, it has also been reported to form complexes with CDK1. It is presumed that CDK2, activated by E-type cyclins and cyclin A, promotes cell-cycle progression from the G1/S boundary through the G2 interval.
At this time, B-type cyclins, in conjunction with CDK1, are responsible for getting cells into and through mitosis. Although mammalian cells express a number of B-type cyclins, only cyclin B1 appears to be essential. Cyclin B1 accumulates through S phase and G2 and then is degraded at the metaphase-anaphase transition. It should be pointed out that the CDK family is extensive and that eukaryotes possess many additional CDKs that ostensibly have nothing to do with cell-cycle regulation.
The blueprint for CDK function through the mammalian cell cycle presented here is based on a large body of experimental evidence and most likely accounts for primary activities at each cell-cycle stage. However, this model does not account for potential redundancy of CDK function. Recently, however, it has been demonstrated that a CDK2-nullizygous mouse is viable, and furthermore, relatively normal.13,14 Investigation of embryonic fibroblasts from these mice has suggested that in the absence of CDK2, CDK1 can carry out the functions normally attributed to CDK2.15 However, the contribution of CDK1 to these functions in unperturbed cells remains to be determined.
Modes of Cyclin-Dependent Kinase Regulation
Because the activity of CDKs is central to cell survival, these enzymes are, of necessity, highly regulated.6 As a result, a number of diverse regulatory mechanisms have evolved to allow for integration of environmental and internal signals (Fig. 6.2). A primary mode of CDK regulation is the availability of activating cyclins, as alluded to previously. For most cell-cycle regulatory CDKs, the relevant cyclins exhibit a distinct temporal program of accumulation and degradation, determining a precise window of CDK activation. Although D-type cyclins tend not to be highly regulated in cycling cells, they are strongly down-regulated as cells exit the cell cycle into a nonproliferative state and then resynthesized in response to mitogen stimulation and cell-cycle re-entry. The genes encoding cyclin E1 and E2 are transcribed periodically late in G1 and up to the G1-S phase transition. This, coupled with ubiquitin-mediated proteolysis of cyclin E in active cyclin E–CDK2 complexes, creates the observed window of cyclin E accumulation from late G1 to mid-S phase.
Like cyclin E, the accumulation of cyclin A is determined by periodic transcription. However, unlike cyclin E, cyclin A remains stable in active CDK2 complexes. The timing of ubiquitin-mediated proteolysis of cyclin A is determined by activation of a protein-ubiquitin ligase known as the anaphase-promoting complex/cyclosome (APC/C) in prometaphase. Thus, the window of cyclin A accumulation is from the G1-S transition until early in mitosis. Finally, B-type cyclin accumulation is also linked to periodic transcription. In this case, transcription begins in late S phase and persists through G2. Similarly to cyclin A, B-type cyclins are targeted for ubiquitin-mediated proteolysis by the APC/C during mitosis, although their disappearance occurs slightly later in mitosis than that of cyclin A.
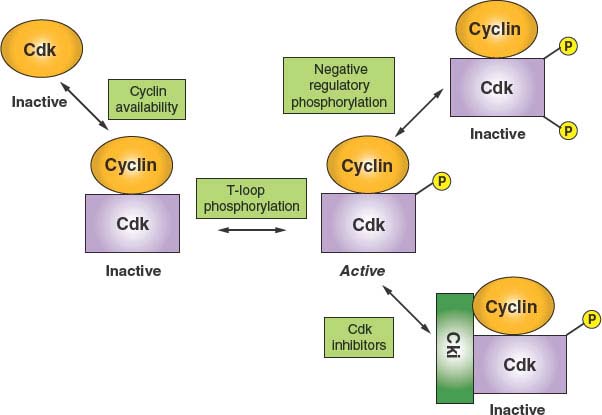
FIGURE 6.2 Principles of cyclin-dependent kinase (CDK) regulation. Because CDK catalytic subunits are inactive when unbound to cyclins, the first level of regulation is through the expression and availability of cyclins. The second level of regulation appears to constitute a housekeeping function in that cyclin binding stimulates an essential phosphorylation of a threonine within a structural feature known as the T loop. Binary complexes with phosphorylated T loops are active. Such active kinase complexes can be subjected to negative regulation in two ways. Additional phosphorylation events on threonine 14 and tyrosine 15 (or the equivalent residues) render the kinase inactive. In addition, the formation of ternary complexes with CDK inhibitory proteins (Ckis) promotes an inactive state. One class of Cki, INK4, specific for CDK4 and CDK6, can inhibit by forming binary complexes directly with the CDK.
It is interesting to note that periodic transcription of cyclins E, A, and B mRNAs relies primarily on negative regulation. For cyclin E, an element known as CERM (cyclin E repressor module) binds a repressor complex containing the repressive member of the E2F family of transcription factors, E2F4, as well as the Rb-related protein p107 and a histone deacetylase.16 Inactivation of the repressive complex in late G1 via phosphorylation of p107 by CDK4/6 allows the constitutive transcription factor, Sp1, to drive cyclin E transcription. Transcription of cyclin A and B mRNAs is similarly regulated. In this case, the repressor element is known as cell cycle genes homology region (CHR)17 but the corresponding repressor complex has not yet been well characterized. However, once repression is relieved, transcription of both cyclin A and B mRNAs is driven the constitutive transcription factor, NF-Y.
A second important mode of CDK regulation is by phosphorylation. CDKs require an activating phosphorylation on a structural feature designated the T loop. Phosphorylation induces a movement of the T loop that has global effects on CDK structure, including an increase in CDK-cyclin contacts and changes in the substrate-binding site.18 In most if not all instances, T-loop phosphorylation appears to constitute a housekeeping function that occurs concomitant with cyclin binding. However, negative regulatory phosphorylation of CDKs is a highly dynamic process. Proper cell-cycle regulation of CDK1, in particular, requires phosphorylation on two residues within the N-lobe, adjacent to the ATP-binding site: threonine 14 and tyrosine 15. During the normal course of the cell cycle, as cyclin B–CDK1 complexes accumulate, they are immediately phosphorylated at these sites and thereby kept inactive. This allows stockpiling of the large numbers of cyclin B–CDK1 complexes required for efficient entry into mitosis and maintaining them in an inactive state during late S phase and G2. At the G2/M-phase boundary, there is concerted dephosphorylation of these residues, causing cells to advance rapidly into mitosis. Although CDK2 and CDK4 have also been reported to undergo negative regulatory phosphorylation at the homologous residues, the function(s) of this regulation are not as clear-cut (but see “DNA Damage Checkpoints”).
A third mode of CDK regulation is through the action of inhibitory proteins that can form either binary complexes with CDKs or ternary complexes with cyclin-CDK dimers. These exist in three major families. The INK4 family consists of four members (p15, p16, p18, and p19). All are composed of a series of conserved structural motifs known as ankyrin repeats and they specifically target CDK4 and CDK6. The mechanism of action of these inhibitors is to bind the CDK subunit and, by causing a rotation of the N-lobe relative to the C-lobe, constraining the kinase in an inactive conformation and, in addition, precluding cyclin binding.19 The Cip/Kip family consists of three members in mammals: p21Cip1, p27Kip1, and p57Kip2. All contain a conserved amino terminal cyclin-CDK–binding inhibitory domain and a divergent C-terminal domain possessing other less well-characterized functions. Although these have been characterized primarily as potent inhibitors of CDK2 and have more recently been shown to also be effective CDK1 inhibitors, the case for inhibition of CDK4 and CDK6 is less certain. Whereas Cip/Kip inhibitors are clearly capable of inhibiting CDK4 and CDK6 at high concentration, it is not clear that these conditions are met in vivo, and the situation is further complicated by the finding that Cip/Kip inhibitor binding is actually required to provide a chaperonin or assembly function for the efficient formation of active cyclin D–CDK4 complexes.20 In the case of cyclin A–CDK2, where structural studies have been carried out, it appears that the Cip/Kip inhibitors first anchor via a high-affinity interaction with the cyclin.21 This then allows the inhibitor polypeptide to invade and deform the N-lobe, thus interfering with ATP binding and catalysis.16 The final class of inhibitors consists of two members of the pRb protein family, p107 and p130. Although these proteins have well-characterized functions as transcriptional inhibitors, they also are potent cyclin E/A–CDK2 inhibitors. p107 and p130 each contain cyclin-binding and CDK-binding sites that collaborate to confer inhibitory activity.
A final mode of CDK regulation is via control of nuclear import/export. This level of regulation is most obvious for cyclin B–CDK1 complexes, which are kept out of the nucleus via active nuclear export until late G2, when phosphorylation inactivates cis-acting nuclear export signals allowing nuclear accumulation.22 Sequestration of cyclin B–CDK1 in the cytoplasm is a redundant mechanism, along with negative regulatory phosphorylation of CDK1, for preventing premature phosphorylation of mitotic targets.
INDUCTION OF CELL-CYCLE PHASE TRANSITIONS
The cell cycle is composed of two action phases, S phase and M phase, in which the genetic material is duplicated and the components of a mother cell are divided into two daughter cells, respectively. The intervening phases, G1 and G2, are thought to exist primarily to allow time for cell growth and for regulatory inputs. Therefore, from the point of view of regulatory theory, cell proliferation is controlled operationally at two key transitions: that between G1 and S phase and that between G2 and M phase. The important characteristic of these two transitions is that, once initiated based on integration of regulatory signals, they must be executed decisively to maintain genetic and genomic integrity. This is accomplished by using a combination of positive and negative modulators to set up the equivalent of a molecular capacitor.
In cycling mammalian cells, the programmed accumulation of cyclins E and A via transcriptional induction provides the positive impetus for the G1-S phase transition. However, these kinases are kept in check by the action of Cip/Kip family inhibitors. If the internal and external environments are permissive for proliferation, the continued accumulation of cyclins will eventually titrate the inhibitors, allowing the latter to be phosphorylated by free cyclin-CDK complexes. Phosphorylation then marks these inhibitors as targets of ubiquitin-mediated proteolysis. The concerted destruction of CDK inhibitors and concomitant activation of the entire pool of CDK complexes assure that the transition into S phase is rapid and irreversible.
Although the details of its regulation are somewhat different, the strategy underlying control of the G2-M transition is similar. Cyclin B–CDK1 complexes accumulate starting near the end of S phase but are held in check not by CDK inhibitors but by negative regulatory phosphorylation of CDK1. This phosphorylation on threonine 14 and tyrosine 15 is carried out by kinases Wee1 and Myt1. Entry into M phase is signaled by the rapid dephosphorylation of T14 and Y15, resulting in activation of CDK1. This dephosphorylation is carried out by specialized protein phosphatases, CDC25B and CDC25C. The concerted dephosphorylation of CDK1 depends on activation of CDC25 isoforms by phosphorylation, as well as ubiquitin-mediated proteolysis of Wee1, also in response to phosphorylation. Although the initial activation of CDC25 isoforms is thought to be carried out by other protein kinases, such as Plk1, CDC25B and C are also activated by cyclin B–CDK1, establishing a positive feedback loop. These positive feedback dynamics leading to the simultaneous activation of a large accumulated pool of cyclin B–CDK1 assures that entry into mitosis is decisive. The turnover of Wee1 enforces irreversibility. Because entry into mitosis involves dismantling many of the cell’s components and organelles, as well as construction and use of a complex apparatus for segregating the cell’s genetic material, mitosis is a period of particular vulnerability, and therefore it is important that this transition and subsequent events be carried out rapidly and efficiently.
An important secondary transition that occurs within M phase is that between metaphase and anaphase (Fig. 6.3).23 To preserve genomic integrity, all duplicated chromosomes must be aligned along the cell’s equator and properly attached to microtubules of the mitotic spindle. The trigger for separation of sister chromatids and their movement to opposite poles of the cell is the activation of the protein-ubiquitin ligase APC/C. This is achieved via CDK1 activation, but more importantly by the binding of a key cofactor, CDC20, whose availability is linked to the proper attachment of chromosomes and the integrity of the spindle. The targets of the APC/C are a protein known as securin as well as cyclin B, both of which inhibit a specialized protease, separase. The key target of separase is a complex that binds sister chromatids together: cohesin. Cleavage of the Scc1 subunit of cohesin leads to a rapid execution of anaphase. It is the ability to stockpile a large pool of inactive securin-separase complexes that can be rapidly mobilized by irreversible proteolysis of securin and cyclin B that allows for a rapid irreversible metaphase-anaphase transition.
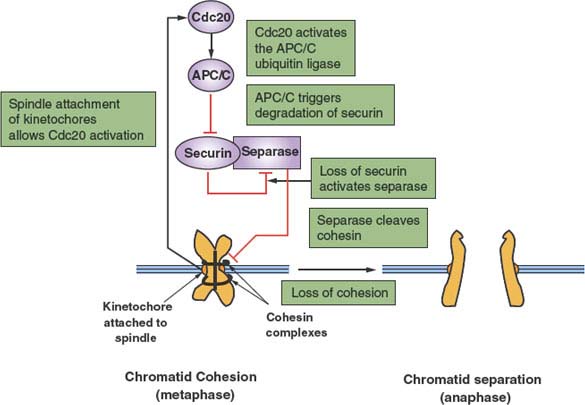
FIGURE 6.3 Regulation of the metaphase-anaphase transition. After replication, paired sister chromatids are held together by a complex of proteins, known collectively as cohesin, thus preventing anaphase. However, once paired chromatids have formed bivalent attachments to a functional mitotic spindle, inhibition of CDC20 mediated by the chromosome kinetochores is relieved. CDC20 is an essential cofactor of the protein-ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C), which now becomes active. The primary initial target of the APC/C is securin, an inhibitor of the protease separase. Ubiquitin-mediated proteolysis of securin releases separase to cleave the cohesin subunit Scc1. This event releases paired chromatids from cohesion, thereby allowing anaphase to proceed.
UBIQUITIN-MEDIATED PROTEOLYSIS
It is becoming increasingly evident that much of the regulation of cell-cycle phase transitions depends on ubiquitin-mediated proteolysis.24 Ubiquitin is a 76-amino acid polypeptide that can be covalently linked to lysines of other proteins via the formation of an isopeptide bond with its C-terminal carboxylate. Additional ubiquitin molecules can then be attached to the lysines of already conjugated ubiquitin to form polyubiquitin chains. Polyubiquitylated proteins are usually targeted for rapid proteolysis by a large multisubunit protease known as the proteasome. The enzymes that transfer ubiquitin to target proteins are known as protein-ubiquitin ligases. From the perspective of cell-cycle control, two families of protein-ubiquitin ligases have predominant roles. The first family, SCF (Skp1-Cullin–F-box protein), specifically targets proteins that are marked for destruction by phosphorylation. This allows degradation of specific proteins to be regulated at a separate level from the protein-ubiquitin ligase itself, which can be expressed constitutively and used to target a large number of substrates independently. SCF ligases consist of three invariant core components and one of a number of specificity factors (F-box proteins) that recognize phosphorylated substrates. A few notable examples of SCF protein-ubiquitin ligases are SCFSkp2 (containing the F-box protein Skp2), which targets p2725 and p130,26 and SCFCDC4 (containing the F-box protein CDC4, also known as FBXW7), which targets cyclin E.27–29 The second family of protein-ubiquitin ligases that is critical for cell-cycle control is known collectively as the APC/C. The APC/C is a large complex consisting of 12 core subunits and 1 of 2 specificity factors, CDC20 and CDH1. Unlike SCF ubiquitin ligases, targeting of substrates by the APC/C is determined by ligase activation rather than substrate activation. APCCDC20 is active from metaphase until the end of mitosis as a result of periodic accumulation and degradation of CDC20 itself. APCCDH1, on the other hand, which is negatively regulated by CDK-mediated phosphorylation, is activated on mitotic exit and during the subsequent G1 interval when CDKs are inactive. In this manner, important mitotic targets, such as cyclin A, cyclin B, and securin (as well as many others), are degraded during mitosis and prevented from reaccumulating during the subsequent G1 interval.
REGULATION OF THE CELL CYCLE
To preserve organismic function and integrity, the cell cycle must be regulated at a number of levels. These include entry into and exit from proliferation mode, coordination of cell-cycle events, and specialized responses that increase the probability of surviving a variety of environmental and internally generated insults.
Quiescence and Differentiation
The most fundamental aspect of cell-cycle control is the regulation of entry and exit. For mammalian cells, the decision to enter or exit the proliferative mode is based on environmental signals such as mitogens, growth factors, hormones, and cell-cell contact, as well as on internal differentiation programs. If the state of cell-cycle exit is reversible, it is referred to as quiescence. If it is in the context of terminal differentiation, cell-cycle exit may merely be one component of a differentiation program. Although cells entering quiescence and postmitotic differentiation vary from each other in many respects, from the perspective of cell-cycle control, they have much in common. First, cell-cycle exit is usually associated, at least initially, with an accumulation of G1/S CDK inhibitors. Members of the INK4 family, targeting CDK4 and CDK6 and members of the Cip/Kip family, as well as the Rb-related protein p130, all targeting CDK2, are up-regulated. This causes accumulation of cells in G1, from where cell-cycle exit can occur. Next, or simultaneously, the positive cell-cycle machinery is dismantled by down-regulation of CDKs and cyclins, primarily at the transcriptional level. In the case of quiescence, cell-cycle exit is paralleled by a reduced rate of protein synthesis, indicative that cells have entered a resting state.
Entry into and exit from quiescence are mediated largely by growth factors and mitogens that interact with cell surface receptors. These in turn are linked to intracellular signaling cascades that up-regulate the rate of protein synthesis as well as the transcription of genes that promote proliferation, such as those encoding CDKs and cyclins. The two best-characterized signaling pathways in this context are the mitogen-activated protein kinase/extracellular signaling–regulated kinase pathway30 and the phosphoinositide 3 (PI3) kinase/AKT pathway,31 shown in Figure 6.4.. Whereas the mitogen-activated protein kinase/extracellular signaling–regulated kinase pathway tends to stimulate expression of genes required for proliferation, the PI3-kinase/AKT pathway primarily stimulates protein synthesis and growth but also affects key cell-cycle regulatory proteins. Just as the presence of growth factors and mitogens stimulates these pathways, promoting cell-cycle entry, their removal shuts down these pathways, promoting quiescence. This is the basis for the reversibility of the quiescent state.
Antimitogenic Signals
An important aspect of control of cell division in mammals is antimitogenic signaling. Just as mitogens and growth factors bind to transmembrane receptors and use signal transduction pathways and downstream transcriptional programs to stimulate proliferation, parallel systems antagonize proliferation. The classic example of an antimitogenic signal is the effect of transforming growth factor-β (TGF-β) on epithelial cells (Fig. 6.5).32 TGF-β, a cytokine, binds to a specific heterodimeric transmembrane receptor that, when occupied by ligand, phosphorylates a class of transcription factors, known as SMADs. These phosphorylated SMADs heterodimerize with non–receptor-interactive SMADs and translocate to the nucleus, where they complex with DNA-binding transcription factors and coactivators to transactivate specific genes. Relevant to cell-cycle regulation, stimulation of the TGF-β signaling pathway promotes transcription of the gene encoding p15. p15 is an INK4 class CDK inhibitor that specifically inactivates CDK4 and CDK6. However, the effects of p15 accumulation on cell-cycle regulators are more global than inhibition of CDK4/6.33 INK4 inhibitors such as p15 have a secondary effect of displacing a pool of the Cip/Kip inhibitor p27 from cyclin D–CDK4/6 complexes, allowing it to then target and inactivate cyclin E–CDK2 and cyclin A–CDK2 (Fig. 6.5). Thus, exposure of epithelial cells to TGF-β has the effect of inhibiting G1 and S phase CDK activities, thereby causing G1 arrest.
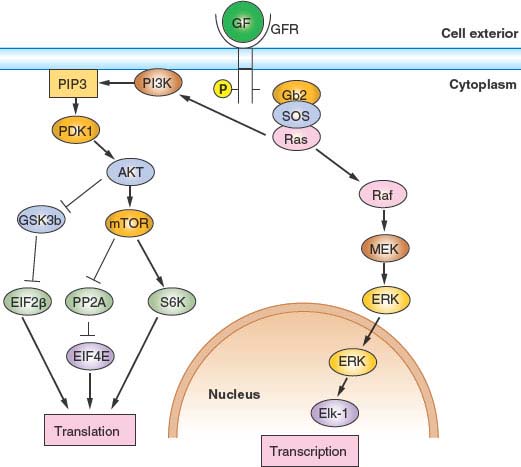
FIGURE 6.4 Growth factor (GF)/mitogen stimulator in response to occupancy of many GF receptors (GFRs) by ligand depends on the small guanosine triphosphatase transducer Ras. Receptor activation leads to phosphorylation of the receptor cytoplasmic domain. The phosphorylated receptor assembles a complex that includes Ras and its activated nucleotide exchange factor, son of sevenless (SOS), leading to activation of Ras. Activated Ras can then stimulate two important signal transduction pathways: the extracellular signaling–regulated kinase (ERK) pathway and the phosphoinositide 3 kinase (PI3K) pathway. Activated Ras stimulates the protein kinase activity of Raf, activating a protein kinase cascade consisting of Raf, MEK, and ERK. Activated ERK then translocates into the nucleus, where it phosphorylates and activates transcription factors, notably Elk-1. Genes important for growth and division are then transcribed. Activated Ras also stimulates PI3K activity, leading to the accumulation of phosphatidylinositol 3,4,5-triphosphate. This in turn stimulates the protein kinase activity of phosphoinositide-dependent kinase 1 (PDK1), activating a protein kinase cascade consisting of PDK1, AKT, and mTOR. Activation of this signal transduction pathway has the effect of stimulating translation and growth. AKT phosphorylates and inhibits the protein kinase glycogen synthase kinase 3 (GSK3β), thereby activating EIF2B required for translational initiation. mTOR phosphorylates and inhibits the protein phosphatase PP2A, thereby activating EIF4E, also required for translational initiation. Finally, mTOR phosphorylates and activates pp70S6 kinase, which in turn phosphorylates and activates ribosomal subunit S6.
Interestingly, in many cancers of epithelial origin, the response to TGF-β has been abrogated, suggesting that this and similar response pathways have an important role in maintaining control of proliferation. Interferons comprise another class of cytokines that have antiproliferative effects on many cell types. Although the receptors used and signaling pathways are distinct from those used by TGF-β, the ultimate effects on the cell-cycle machinery are similar: up-regulation of CDK inhibitors and down-regulation of cyclins.
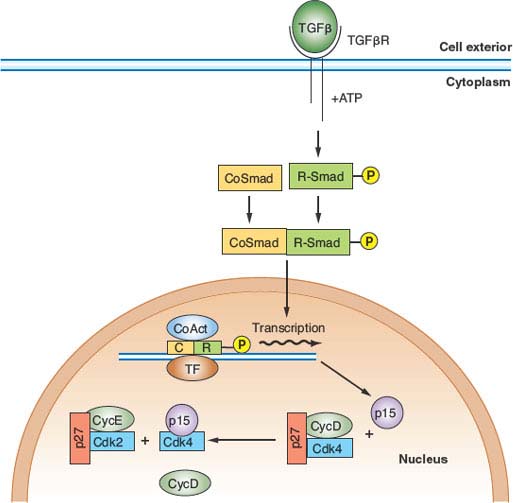
FIGURE 6.5 Transforming growth factor-β (TGF-β) antimitogenic pathway. Occupancy of the heterodimeric TGF-β receptor by ligand leads to phosphorylation of a class of transcription factor known collectively as R-SMADs. Phosphorylated R-SMADs then dimerize with nontargeted cofactors known as CoSMADs and translocate to the nucleus. There the R-SMAD/CoSMAD dimers complex with DNA-binding transcription factors and transcriptional coactivators to stimulate transcription of specific genes. One of the key targets of TGF-β signaling is the gene encoding p15/INK4b, a CDK4/6 inhibitor. p15, by binding to CDK4 and CDK6, inhibits these kinases and displaces a large pool of CDK4/6-bound p27, which is then free to inhibit CDK2 complexes. The result is G1 arrest. ATP, adenosine triphosphate; TF, transcription factor; CoAct, coactivator.
Checkpoints
Cells are constantly faced with insults, resulting in damage that can threaten their survival. These insults can be generated internally as chemically active by-products of metabolism or can originate in the external environment; for example, chemical agents or radiation. As a result, mechanisms have evolved to remove damaged molecules and make necessary repairs. In instances in which cell-cycle progression would be harmful or catastrophic before repair of damage, further mechanisms have evolved to delay progression pending repair. These are called cell-cycle checkpoints.34 The necessity of checkpoints can be easily envisioned for genotoxic agents. Cells are particularly susceptible to the harmful effects of DNA damage at two points in the cell cycle: S phase and M phase. Unrepaired DNA damage poses a number of problems for cells undergoing DNA replication. Chromosomal lesions present physical barriers to replication forks. Replication that does traverse regions of unrepaired DNA damage is likely to be error-prone, resulting in accumulation of mutations. Likewise, segregation of severely damaged chromosomes at mitosis might lead to loss of genetic information, seriously threatening the survival or integrity of daughter cells. Therefore, cells possess mechanisms for preventing DNA replication and mitosis in response to genotoxic stress. Although the scope of this review does not permit a detailed description of all known checkpoints, those thought to be most basic to cell survival are characterized here.
DNA Damage Checkpoints
Although DNA damage exists in many forms, ranging from chemical adducts to double-strand breaks, they all pose similar problems for proliferating cells. As previously stated, impeded and error-prone DNA replication and loss of genetic material during mitosis are some of the likely consequences in the absence of DNA damage checkpoints. Therefore, cell-cycle progression is blocked at three points: before S phase entry (the G1 DNA damage checkpoint), during S phase (the intra-S phase DNA damage checkpoint), and before M phase entry (the G2 DNA damage checkpoint). Although the responses to different types of DNA damage are not identical, they are similar enough to generalize. DNA damage of various forms is first detected by DNA-bound protein complexes that serve as sensors. In mammalian cells, two related atypical protein kinases that share homology with lipid kinases, ATM and ATR,35 are primary signal transducers that are activated by DNA damage at all points in the cell cycle. A key effector of the G1 and G2 checkpoint responses is a transcription factor known as p53.36,37 In response to DNA damage, p53 is activated and stabilized leading to increased levels. The principal transcriptional target of p53 in the context of the G1 checkpoint is the Cip/Kip inhibitor p21Cip1. The resulting high levels of p21 block CDK2 activity and possibly CDK4 and CDK6 activity, leading to G1 arrest. An additional transcriptional target of p53, GADD45, inhibits CDK1, thereby contributing to the G2 DNA damage checkpoint. Another p53-dependent mechanism contributing to checkpoint-mediated G2 arrest is through transcriptional repression of the genes encoding cyclin B1 and CDK1. This occurs via direct interaction p53 and NF-Y, the positive transcriptional activator of these genes. However, although p53-dependent mechanisms are required for long-term maintenance of arrest, the primary mechanism underlying the immediate G2 DNA damage checkpoint is p53-independent. It involves one of two effector protein kinases known as chk1 and chk2 that have the effect of inhibiting CDC25C,38 which carries out the activating dephosphorylation of CDK1. Therefore, in response to DNA damage, G2 cells accumulate inhibited cyclin B–CDK1 complexes and are incapable of entering into mitosis. The intra-S phase DNA damage checkpoint response appears to be p53-independent but requires the chk1 or chk2 kinases, or both. A key target is CDC25A, responsible for activating CDK2 by dephosphorylation. In response to DNA damage, phosphorylation of CDC25A by chk1 or chk2 leads to its destabilization via ubiquitin-mediated proteolysis and thus the accumulation of inactive CDK2 complexes39 phosphorylated on threonine 14 and tyrosine 15. Because ongoing DNA replication requires the activity of CDK2, DNA synthesis ceases until damage is repaired.
Replication Checkpoint
Under normal circumstances, DNA replication is complete well before the time when the accumulation and activation of cyclin B–CDK1 would drive cells into mitosis. However, through the action of toxins or the rare but finite probability that the duration of S phase will be excessively long, situations can be encountered in which completion of replication extends beyond the normal time of mitotic induction or replication is blocked entirely. Under such circumstances, it is necessary to delay or block entrance into M phase accordingly, as segregation of incompletely replicated chromosomes would be catastrophic, leading to chromosome breaks and/or nondisjunction events. Although the signaling pathways are somewhat different, the replication checkpoint ultimately functions like the G2 DNA damage checkpoint in that mitotic entry is blocked by inhibiting CDC25C via the action of chk1, thus preventing activation of CDK1.
Spindle Integrity Checkpoint
The actual act of division is a dangerous time for a cell. It requires aligning duplicated chromosomes by attaching them via bipolar attachments to the spindle and then separating the chromatids so that each daughter cell gets a full complement. Errors result in aneuploidy, an extremely undesirable outcome. As a result, assembling a mitotic spindle and attaching chromosomes to it are extensively monitored processes. The mechanism of delay at prometaphase or metaphase in response to spindle defects or improper chromosome attachment is referred to as the spindle integrity checkpoint.40 The sensor for this checkpoint consists of a number of proteins that reside at the chromosome kinetochores, sites of spindle microtubule attachment. The target is the essential APC/C cofactor, CDC20. Unattached or improperly attached kinetochores not experiencing an appropriate level of tension indicative of bipolar attachment inhibit CDC20 function. This in turn prevents the ubiquitylation and degradation of the anaphase inhibitors, securin and cyclin B. As a result, cells are prevented from initiating anaphase until all kinetochores are properly attached to a bipolar spindle (Fig. 6.3).
Restriction Point
Cells deprived of an essential nutrient or growth factor are blocked from cell-cycle progression at a point in mid-G1.41 Cells that have already passed this point, termed the restriction point or R, enter into S phase and complete the current cell cycle before arresting in the subsequent G1 interval. In contrast, G1 cells that have not reached the restriction point arrest immediately. The molecular basis for the restriction point has remained elusive. Initially it was thought that passage through the restriction point was a manifestation of G1 CDK activation and/or phosphorylation the pRb family of transcriptional inhibitors. However, more recent work has indicated that CDK activation and pRb phosphorylation occur after passage through the restriction point.42,43 Significantly, most malignant cells do not have a functional restriction point, which presumably helps them evade normal growth control signals.
Senescence
All normal mammalian cells have a finite proliferative lifespan. As cells approach the end of their proliferative capacity, they enter a state referred to as replicative senescence.44,45 Although the reasons for programmed senescence are not known, it has been speculated that restricting cells to a finite number of divisions may be a protective mechanism against malignant growth. Although the rationale for senescence is not known, the mechanism has been largely elucidated, particularly for human cells. It is based on the requirement for a specialized replicase, telomerase, in the replication of the ends of chromosomes known as telomeres. Whereas germ line cells express telomerase, most if not all somatic cells do not. As a result, because of the topology of telomeres and the requirements of conventional DNA replication, progressive telomere shortening or attrition occurs with each cell cycle. Although linear chromosome ends create a discontinuity, which topologically is indistinguishable from a chromosome break, telomere-specific DNA sequences are shielded from the DNA damage checkpoints. However, when sufficient telomere attrition has removed these protected sequences, cells enter into a chronic checkpoint response, which is the molecular basis for senescence. Senescence is characterized by the accumulation of high levels of CDK inhibitors and ultimately permanent G1 arrest. It should be noted that one of the requirements of malignant transformation of cells is to overcome the senescence barrier so as to provide tumor cells with unlimited proliferative capacity.
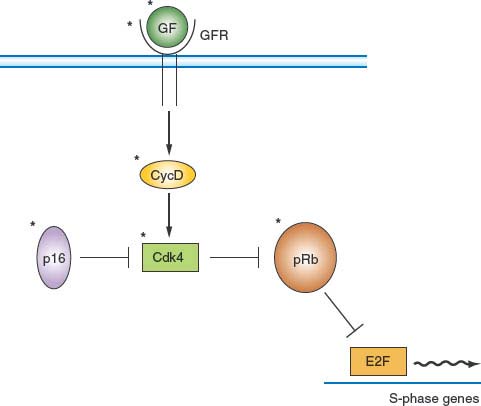
FIGURE 6.6 pRb pathway. pRb is a critical negative cell-cycle regulator that links growth factor (GF) signaling pathways to cell-cycle progression. One of the principal functions of pRb is to interact with E2F family transcription factors and, by recruitment of corepressors, to maintain many genes encoding proteins that are important for cell-cycle progression in a tightly repressed state. GF and mitogen-signaling pathways relieve this repression by stimulating accumulation of D-type cyclins on receptor occupancy. The resulting activation of CDK4 and CDK6 leads to phosphorylation of pRb and concomitant inactivation of its repressive functions. p16 is a CDK inhibitor of the INK4 family that down-regulates this pathway by inhibiting CDK4. It should be noted that all elements marked by an asterisk are found mutated or deregulated, or both, in human cancer. GFs, GF receptors (GFRs), and D-type cyclins are frequently overexpressed or deregulated. p16 is often not expressed or is underexpressed. Mutant versions of CDK4 that cannot bind p16 have been identified in human cancers. Finally, the gene encoding pRb is frequently mutated in cancer.
Regulation of DNA Replication
Entry into S phase is one of the key regulatory points of the cell cycle. The actual triggering of replication is attributed to the activation of CDK2 by cyclins E and A. However, the transcription of a large number of genes whose products are required for DNA replication requires the activity of CDK4 or CDK6, or both, driven by D-type cyclins. Mechanistically, this is based on the function of pRb and related proteins p130 and p107 serving as transcriptional repressors when bound to E2F family transcription factors (Fig. 6.6).8 Phosphorylation by cyclin D–CDK4/6 relieves this repression. Once cells have synthesized all the necessary enzymes and initiated DNA replication, another serious regulatory problem is encountered. To maintain genomic integrity, cells must replicate all genomic sequences only once per cell cycle, necessitating that origins of replication, sites where DNA synthesis begins, are used once during each S phase. This is accomplished by requiring that replication origin preparation and firing are mediated, respectively, by distinct CDK environments.46 Pre-replication complex assembly is triggered by low or absent CDK activity and therefore normally occurs as cells exit mitosis. This process requires the successive loading of proteins, CDC6, ctd1, and six MCM proteins (MCM2–7) to another complex of proteins, known as the origin recognition complex, which marks the origin site. Because of the requirement for low CDK activity, the permissive window for this process extends from the end of mitosis (telophase) until the point in G1 when CDK activity begins to rise. The activation of CDK2 in late G1 has the dual effect of blocking further pre-replication complex assembly and causing DNA replication to initiate at primed origins. The maintenance of high levels of CDK activity (CDK2 followed by CDK1) for the remainder of the cell cycle assures that no new pre-replication complex assembly can occur until the end of mitosis, when CDK levels once again decline, and in doing so restricts origin function to once per cell cycle. Indeed, inhibiting CDK1 activity during G2 or early M phase is sufficient to promote a round of DNA replication without cell division.
CELL CYCLE AND CANCER
Cancer is partly a disease of uncontrolled proliferation. Because the proliferation of cells within an organism is normally tightly controlled by redundant regulatory pathways, it is not surprising that cell-cycle and checkpoint genes are often found misregulated or mutated in cancer. Genes in which mutations give rise to a gain of function or an enhanced level of function, leading to malignancy, are referred to as protooncogenes. Protooncogenes usually encode growth- or division-promoting proteins. Genes that give rise to loss of function mutations that lead to malignancy are referred to as tumor suppressor genes. Tumor suppressor genes usually encode negative regulators of growth and proliferation that protect cells from malignancy. Some cell-cycle genes commonly mutated or misregulated in cancer are listed in Table 6.1. Whereas mutations that create oncogenes tend to be dominant, mutations in tumor suppressor genes are usually recessive. This has led to the two-hit model of carcinogenesis (Fig. 6.7).47 Briefly, recessive mutations occur in tumor suppressor genes but are latent because of the persistence of a wild type allele. The tumor suppressor phenotype, therefore, requires mutation or loss of the second allele, a process known as loss of heterozygosity. Alternatively, the second allele of a tumor suppressor gene can be silenced epigenetically without a direct genetic alteration. A number of genes encoding negative regulators of the cell cycle conform to this two-hit paradigm.
TABLE 6.1
CELL-CYCLE GENES COMMONLY MUTATED OR ALTERED IN EXPRESSION IN HUMAN CANCER
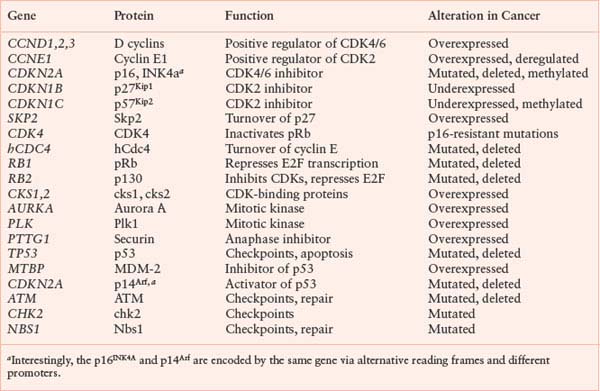
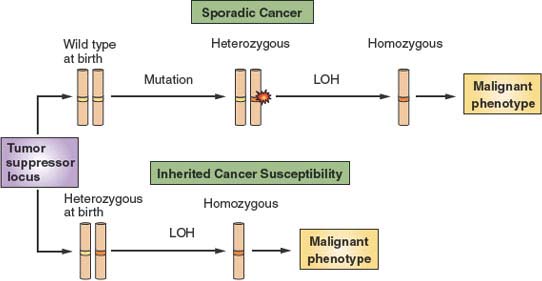
FIGURE 6.7 The two-hit model of tumor suppression. Tumor suppressors are proteins that are thought to provide protection from malignancy. Depicted is a chromosome carrying a tumor suppressor–encoding locus shown in white. At birth, normal individuals carry two wild type alleles (yellow bands) at tumor suppressor loci. Over time, however, spontaneous mutations occur at these loci (red flash) that render one allele nonfunctional (orange band). However, because such mutations are expected to be recessive to the wild type allele that is still present, there is no phenotypic consequence. Over time, additional events can lead to loss of the wild type allele, a phenomenon referred to as loss of heterozygosity (LOH). LOH then provides a tangible contribution to the malignant phenotype. However, because spontaneous mutations at specific loci and specific secondary allelic losses are rare events, malignancy usually develops only after a very long latency period. On the other hand, some individuals are born with inherited tumor suppressor mutations. Because only LOH is then required for expression of the tumor suppressor–null phenotype, cancer with decreased latency and higher penetrance develops in such individuals.
In theory, to achieve uncontrolled cell division, two basic requirements must be met. First, cells need a strong constitutive proliferation signal capable of overriding the environmental and internal restraints on division that normal cells experience. Second, the barrier of senescence needs to be dismantled to render tumor cells immortal. Mutations in a large variety of cell-cycle control and related genes are associated with malignancy, and most of these can be accommodated within this framework. This model of tumorigenesis has been confirmed in rodent tissue culture–based in vitro models. Transfection of primary rodent fibroblasts with individual plasmids programmed to express proteins that promote either growth or immortalization does not result in malignant transformation. However, cotransfection of two plasmids, one in each category, does promote transformation (Fig. 6.8). However, these results need to be interpreted cautiously in the context of human cancer because immortalization of rodent cells in culture most likely does not involve telomeres, which are much longer in rodents than in humans.48 One idea that has emerged is that strong growth signals and other environmental pressures exerted on premalignant cells produce potent stress responses, leading to cell-cycle blockade or cell death.49 Phenotypically, such stress-induced effects on fibroblasts closely resemble those associated with replicative senescence; therefore, this phenomenon has been termed stress-induced senescence (see following discussion). Therefore, genetic alterations are likely required to neutralize these stress responses to immortalize rodent cells. Transformation of human cells requires these same genetic alterations, but also telomere attrition must be reckoned with, requiring additional mutations.
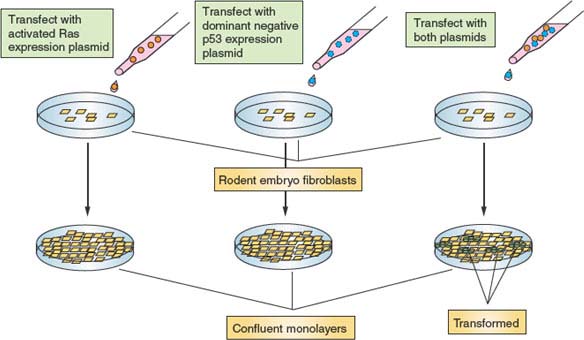
FIGURE 6.8 Malignant transformation requires multiple genetic alterations. Depicted is an in vitro experiment using primary rodent embryonic fibroblasts. Cells transfected with a plasmid programmed to express an activated Ras allele eventually grow to form a confluent monolayer, at which time proliferation ceases because of inhibition mediated by cell-cell contact. Similarly, cells transfected with a plasmid programmed to express a dominant-negative allele of p53 (encoding a protein that can complex with and inactivate endogenous wild type p53) from a confluent monolayer. However, cells transfected simultaneously with both plasmids from a confluent monolayer, out of which grow transformed foci. These piles of cells are no longer subject to the controls that restrict fibroblast proliferation and, as such, resemble cancer cells. The requirement for two perturbations in this system supports a mechanism whereby activated Ras stimulates growth and proliferation, and dominant negative p53 inactivates stress pathways that would cause these cells to have a limited proliferative lifespan.
Alterations in Pathways Affecting Growth and Proliferation
Mutations that regulate cell growth and proliferation can occur at many levels, ranging from cell surface receptor–mediated signaling pathways that control proliferation to elements of the core cell-cycle machinery itself.
Growth and Proliferation Signaling Pathways
Because a large number of receptors and pathways can influence cell proliferation, many mutations in elements of these pathways have been recovered in human malignancies. Only a few examples are cited here. One way to provide a strong constitutive proliferation signal is to overexpress or deregulate growth factor receptors. HER2/neu, a transmembrane tyrosine kinase receptor found on many epithelial cell types, is often overexpressed because of gene amplification in breast and other cancers.50 Presumably the amplitude of proliferation signaling is abnormally high or completely deregulated in such tumors. Similarly, signaling elements downstream of mitogen receptors can be mutated to produce constitutive signaling. Perhaps the best-known example is the case of the Ras family guanosine triphosphatases, which serve as signal transducers for a number of key proliferation pathways. Dominant mutations in Ras isoforms that stabilize the activated state confer strong constitutive proliferation signaling. One of the pathways stimulated by Ras is the PI3-kinase pathway. A PI3 phosphatase, PTEN, normally reverses this phosphorylation, keeping the signal in check. Consistent with this, mutational loss of PTEN similarly to oncogenic mutations in Ras can lead to constitutive signaling contributing to carcinogenesis.
Cell-Cycle Machinery
Signaling pathways that stimulate proliferation impinge on the cell-cycle machinery by stimulating the biosynthesis of D-type cyclins and promoting the degradation of CDK inhibitors. Accumulation of D-type cyclins and concomitant activation of CDK4 and CDK6 have been shown to activate the cell-cycle program primarily by phosphorylation and inactivation of the retinoblastoma protein, pRb, and related proteins p107 and p130. These proteins form potent repression complexes with transcription factors that are critical for S-phase entry and progression, notably the E2F family, effectively blocking cell-cycle progression. In addition, INK4 family inhibitors specifically down-regulate CDK4 and CDK6, buffering their capacity to phosphorylate pRb and related proteins. Virtually all components of this pathway have been found to be misregulated or mutated in cancer to provide a constitutive proliferation signal (proteins with asterisks in Fig. 6.6).51 The genes encoding D-type cyclins are found amplified in a broad spectrum of tumors. On the other hand, the gene encoding the INK4 inhibitor, p16, is mutated and lost in some types of cancer, whereas CDK4 has been found to be mutated so as not to bind p16. In many instances p16, although not genetically altered, is down-regulated at the epigenetic level. The p16 promoter contains a CpG island that is subject to repression via methylation. Finally, pRb is the tumor suppressor on which the so-called two-hit hypothesis was originally formulated. Inherited mutations in the RB gene and subsequent loss of heterozygosity invariably lead to childhood retinoblastoma and eventually other malignancies. However, somatic mutation of RB1 and loss of heterozygosity are found in many sporadic noninherited cancers, underscoring the critical nature of negative cell-cycle regulation by pRb.
Like D-type cyclins, cyclin E is frequently found up-regulated in cancer. The fact that deregulated expression of cyclin E can drive cells prematurely into S phase suggests that cyclin E provides a growth/division stimulus during carcinogenesis. Furthermore, cells from cyclin E nullizygous mice are resistant to malignant transformation in tissue culture models.12 However, other evidence suggests that deregulation of cyclin E may promote carcinogenesis principally by inducing genomic instability rather than by promoting growth (see “Mutations Causing Genetic and Genomic Instability”). Likewise, the CDK2 inhibitor p27Kip1 is often found down-regulated in cancer, although never behaving as a classic tumor suppressor inactivated through mutation and allelic loss. However, as with cyclin E deregulation, it is not clear whether low p27 levels have an impact on carcinogenesis by promoting growth or genomic instability. While the CDK-inhibitory functions of p27 are restricted to the nucleus, hyperphosphorylation leads to cytoplasmic translocation, where non–CDK-bound p27 promotes cell migration. This may explain why p27 is rarely deleted in cancer, as cytoplasmic functions may be important for invasion and metastasis.52
Alterations in Pathways Affecting Senescence
In addition to a constitutive growth stimulus that overrides natural restraints, tumors need to have the capacity for unlimited proliferation. Normally, the limited lifespan of somatic cells imposed by the process of replicative senescence constitutes a natural barrier to tumorigenesis. Therefore, genes that mediate senescence are commonly mutated in cancer. However, the issue of senescence is complicated by functional overlap between senescence pathway genes and oncogenic stress pathway genes that also require inactivation.48,49 Because senescence is a result of checkpoint responses to acute telomere attrition, genes that encode DNA checkpoint signaling elements and transducers are targeted. One of the most commonly mutated genes in human cancer encodes the checkpoint effector p53.36 Inherited mutations in TP53, the gene encoding p53, confer a syndrome known as Li-Fraumeni characterized by early-onset cancer.53 However, the majority of sporadic cancers are also mutated at the p53 locus. The role of p53 mutation in cancer as a promoter of immortalization is supported by the finding that cells from p53 nullizygous mice are immortal.54 However, this conclusion is complicated by the fact that p53 is central to cellular stress responses that also require inactivation during malignant transformation, and as previously stated, telomere attrition is not likely to be a significant issue for immortalization in mice. Nevertheless, an observation supporting the idea that checkpoint genes likely to be triggered by telomere attrition are targeted to immortalize premalignant cells is that chk2, a signaling element of the DNA damage checkpoint response, is mutated in a subset of patients with Li-Fraumeni syndrome53 rather than p53.
Mutation of the gene encoding Nbs1, required for activation of chk1 and chk2 kinases, is also associated with a hereditary cancer syndrome,55 Nijmegen disease, as well as sporadic cancers, although the interpretation of this result is complicated by the fact that Nbs1 is also involved in DNA damage repair (see “Mutations Causing Genetic and Genomic Instability”). However, the most direct strategy to bypass senescence is to induce directly the expression of telomerase in somatic cells. c-Myc, a transcription factor linked to stimulation of proliferation, has also been shown to be a positive regulator of the gene encoding telomerase reverse transcriptase (hTERT), the catalytic subunit of telomerase.56 This may explain the high frequency of human tumors exhibiting c-Myc amplification or overexpression, or both. However, there appear to be a number of different mutational targets that can lead to derepression of the hTERT gene.57
Mutations Neutralizing Stress Responses
Abnormally strong growth and proliferation signals provoke antagonistic stress responses leading to cell-cycle arrest or cell death. For example, it has been observed that expression of mutationally activated Ras alleles in nontransformed human fibroblasts leads to a cell-cycle arrest phenotype that closely resembles replicative senescence. As stated in “Alterations in Pathways Affecting Senescence,” cellular stress responses are intimately related to checkpoint responses. Therefore, it is difficult to clearly categorize mutations that affect both. An example is p53, which is required for DNA damage checkpoint responses but also is a key effector of cellular stress responses.37,48,49 In the case of activated Ras previously cited, the stress-activated MAP kinase pathway promotes phosphorylation and activation of p53 leading to cell-cycle arrest. Therefore, mutations that directly or indirectly inactivate p53 can promote oncogenesis by bypassing stress-dependent cell-cycle arrest or cell death. Murine double-minute gene-2, which is frequently amplified and overexpressed in human cancer, promotes turnover of p53, consistent with a role in neutralizing stress responses.58 Conversely, p14Arf, a protein that stabilizes p53 by antagonizing murine double-minute gene-2, is frequently found mutated or underexpressed in cancer.58 Indeed, the p53 pathway is so frequently inactivated in human cancer most likely because loss of p53 function simultaneously antagonizes stress pathways and helps override cellular responses to telomere attrition. pRb may have a parallel function in maintenance of senescence, as loss of pRb has recently been shown to cause fibroblasts rendered senescent by expression of activated Ras to undergo DNA replication59. Thus, mutation of RB in cancer may contribute to escape from oncogene-induced senescence.
Mutations Causing Genetic and Genomic Instability
The pathway to malignancy minimally requires several mutations. In the case of tumor suppressor mutations, secondary genetic events mediating allelic loss are necessary. Therefore, any mutation that itself can confer genetic or genomic instability, or both, is likely to promote carcinogenesis.60,61 Mutations in genes required for DNA repair result in a mutator phenotype linked to hyperaccumulation of secondary mutations. In this context, strong association between mutation of the gene encoding Nbs1, which is required for efficient DNA repair as well as checkpoint signaling, and carcinogenesis is easily understood.55 Similarly, the association between mutation of components of the spindle integrity checkpoint, such as Bub1, and carcinogenesis can be rationalized.62 Cells defective in this checkpoint experience deregulated mitosis, leading to chromosome instability and ultimately aneuploidy. In principle, aneuploidy potentiates amplification at oncogenic loci and allelic losses at tumor suppressor loci.
An interesting link between the core cell-cycle machinery and genomic instability is the case of cyclin E. Cyclin E is found overexpressed and deregulated in a broad spectrum of malignancies.63 Although this correlation might be interpreted in the context of simply promoting proliferation, experiments on cells in culture have revealed that deregulation of cyclin E expression causes chromosome instability leading to aneuploidy and polyploidy.64 This occurs because expression of cyclin E at inappropriate times in the cell cycle leads to impairment of DNA replication as well as of mitosis. Therefore, one possible role that cyclin E might play in promoting oncogenesis is to accelerate loss of heterozygosity at tumor suppressor loci. This was tested in a transgenic mouse mammary carcinogenesis model and, consistent with this idea, cyclin E deregulation led to accelerated loss of heterozygosity at the TP53 locus (encoding p53), which correlated with higher tumor incidence.65 Interestingly, an essential component of the ubiquitin ligase responsible for cell cycle–dependent targeting of cyclin E for proteolysis, hCDC4 (also known as FBXW7), is often found mutated in cancer,27,29,66 and its deletion has been shown to also cause genomic instability in cultured cells.67 Thus, genetic alterations that interfere with proper regulation of cell-cycle machinery have the potential of affecting not only the cell cycle itself, but also the genetic and genomic integrity of the cell.
MICRORNAs, THE CELL CYCLE, AND CANCER
Although discovered in the nematode Caenorhabditis elegans many years ago, the importance of microRNAs (miRs) in mammalian cells and human cancer has only recently begun to be appreciated.68,69 These small RNAs target specific mRNAs via degradation or inhibition of translation. They confer unique regulatory possibilities in that they usually target several different mRNAs simultaneously, allowing for coordination of multiple pathways. At least five groups or clusters of miRs have been shown to target mRNAs encoding cell-cycle regulatory proteins. The miR-15a/16 cluster targets cyclin E1, cyclin D1, and cyclin D3, as well as CDK6. Not surprisingly, this miR cluster has been shown to have tumor suppressive functions in several different types of cancer. The miR-17/20 cluster, which targets cyclin D1 and E2F transcription factors, and let-7, which targets cyclin D1, cyclin D3 cyclin A, CDK4, and CDK6, have been also been shown to be tumor suppressive. Conversely, the miR-221-222 cluster and mir-21, which target a number of CDK inhibitors, has oncogenic properties. Understanding of the regulation and roles of miRs in normal cell function and cancer etiology is largely incomplete, and filling in the gaps poses a significant research challenge for the coming years.
THE CELL CYCLE AND CANCER THERAPY
Because cancer cells must proliferate, essential cell-cycle proteins have been suggested as targets for therapeutic exploitation. Notably, CDKs have been extensively screened for small-molecule inhibitors, some of which are in clinical trials. It is too early, however, to judge the efficacy of this approach beyond its success using in vitro models. An alternative approach being explored is to develop agents that undermine checkpoint responses. The presumption is that cancer cells, because of their highly proliferative state, might be more susceptible to loss of essential controls. This idea remains to be confirmed. However, it is noteworthy that many therapeutic approaches currently use compounds that normally trigger checkpoint responses, such as genotoxic agents or spindle poisons. It is assumed that these treatments are effective because tumor cells are actually impaired in their defensive checkpoint responses. An interesting approach that initially showed promise in model systems but has proven disappointing in clinical trials, uses the fact that a large percentage of malignancies are defective for p53 function in order to evade checkpoint and stress responses. A common human lytic virus known as adenovirus, expresses an essential gene, E1B p55K, specifically to down-regulate p53 in order to allow a productive infection. Oncolytic adenoviruses have therefore been engineered to not express E1B p55K.70 These adenoviruses are harmless to normal cells but can productively infect and lyse p53-defective tumor cells in tissue culture and mouse xenograft models. However, technical issues such as low tumor infectivity, rapid viral clearance, and neutralizing immune responses in clinical trials have limited the efficacy of this approach.70 On the other hand, if new generations of oncolytic viruses that circumvent these problems can be developed, this may constitute one of the more promising new therapeutic approaches.
Circadian rhythms may present another interesting link between cancer therapeutics and the cell cycle. Although circadian rhythms regulate virtually every aspect of human physiology, it is clear that cell-cycle regulatory gene expression and the cell cycle itself are entrained to the day-night cycle.71 It has also been shown that tumor cells have largely lost this regulation. This difference, therefore, may be exploitable therapeutically. Indeed, the tolerance and efficacy of a variety of genotoxic chemotherapeutic agents varies with time of day, suggesting a possible link between the time of administration and the cell-cycle position of cells in healthy tissues. More detailed understanding of circadian regulation of the cell cycle may provide an avenue for optimization of current therapeutics.
Selected References
The full list of references for this chapter appears in the online version.
2. Johnson RT, Rao PN. Mammalian cell fusion: induction of premature chromosome condensation in interphase nuclei. Nature 1970;226:717.
3. Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature 1970; 225:159.
4. Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool 1971;177:129.
5. Hartwell LH, Culotti J, Pringle JR, et al. Genetic control of the cell division cycle in yeast. Science 1974;183:46.
6. Harper JW, Adams PD. Cyclin-dependent kinases. Chem Rev 2001;101:2511.
7. Jeffrey PD, Russo AA, Polyak K, et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995; 376:313.
8. Stevens C, La Thangue NB. E2F and cell cycle control: a double-edged sword. Arch Biochem Biophys 2003;412: 157.
9. Stiegler P, Giordano A. The family of retinoblastoma proteins. Crit Rev Eukaryot Gene Expr 2001;11:59.
10. Ohtsubo M, Roberts JM. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science 1993;259:1908.
11. Resnitzky D, Gossen M, Bujard H, et al. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol 1994;14:1669.
12. Geng Y, Yu Q, Sicinska E, et al. Cyclin E ablation in the mouse. Cell 2003;114:431.
13. Berthet C, Aleem E, Coppola V, et al. CDK2 knockout mice are viable. Curr Biol 2003;13:1775.
14. Ortega S, Prieto I, Odajima J, et al. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 2003;35:25.
18. Russo AA, Jeffrey PD, Pavletich NP. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat Struct Biol 1996;3:696.
19. Russo AA, Tong L, Lee JO, et al. Structural basis for inhibition of the cyclin-dependent kinase CDK6 by the tumour suppressor p16INK4a. Nature 1998;395:237.
20. Cheng M, Olivier P, Diehl JA, et al. The p21(Cip1) and p27(Kip1) CDK inhibitors are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 1999; 18:1571.
24. Reed SI. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol 2003;4:855.
25. Carrano AC, Eytan E, Hershko A, et al. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999;1:193.
26. Tedesco D, Lukas J, Reed SI. The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev 2002;16: 2946.
27. Strohmaier H, Spruck CH, Kaiser P, et al. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001;413:316.
28. Koepp DM, Schaefer LK, Ye X, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001;294:173.
30. Davis RJ. Transcriptional regulation by MAP kinases. Mol Reprod Dev 1995;42:459.
31. Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia 2003;17:590.
32. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003;113:685.
34. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science 1996;274:1664.
35. Yang J, Yu Y, Hamrick HE. ATM, ATR, and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis 2003;24:1571.
36. Vousden KH. Activation of the p53 tumor suppressor protein. Biochim Biophys Acta 2002;1602:47.
37. Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001;20:1803.
38. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003;3:421.
39. Sorensen CS, Syljuasen RG, Falck J, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003;3:247.
40. Allshire RC. Centromeres, checkpoints and chromatid cohesion. Curr Opin Genet Dev 1997;7:264.
41. Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle 2002;1:103.
42. Ekholm SV, Zickert P, Reed SI, et al. Accumulation of cyclin E is not a prerequisite for passage through the restriction point. Mol Cell Biol 2001;21:3256.
43. Martinsson HS, Starborg M, Erlandsson F, et al. Single cell analysis of G1 check points—the relationship between the restriction point and phosphorylation of pRb. Exp Cell Res 2005;305:383.
44. Smith JR, Pereira-Smith OM. Replicative senescence: implications for in vivo aging and tumor suppression. Science 1996;273:63.
45. Harley CB, Sherwood SW. Telomerase, checkpoints and cancer. Cancer Surv 1997;29:263.
46. Woo RA, Poon RY. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2003;2:316.
47. Knudson AG Jr. Hereditary cancer. JAMA 1979;241:279.
49. Schmitt CA. Cellular senescence and cancer treatment. Biochim Biophys Acta 2007;1775:5.
51. Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta 2002;1602:73.
52. Lee J, Kim SS, The function of p27KIP1 during tumor development. Ex. Mol Med 2009;41:765.
53. Varley J. TP53, hChk2, and the Li-Fraumeni syndrome. Methods Mol Biol 2003;222:117.
58. Zhang Y, Xiong Y. Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth Differ 2001; 12:175.
60. Loeb KR, Loeb LA. Significance of multiple mutations in cancer. Carcinogenesis 2000;21:379.
61. Vessey CJ, Norbury CJ, Hickson ID. Genetic disorders associated with cancer predisposition and genomic instability. Prog Nucleic Acid Res Mol Biol 1999;63:189.
63. Donnellan R, Chetty R. Cyclin E in human cancers. FASEB J 1999;13:773.
64. Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature 1999;401:297.
68. Migliore C, Giordano S. MiRNAs as new master players. Cell Cycle 2009;8:2185.
69. Yu Z, Baserga R, Chen L, et al. microRNA, cell cycle and human breast cancer. Am J Pathol 2010;176:1058.
71. Levi F, Alper O, Dulong S, et al. Circadian timing in cancer treatments. Annu Rev Pharmacol. Toxicol 2010;50:377.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


