FIGURE 24.1 Genetic susceptibility to breast cancer. Familial breast cancer comprises approximately 20% to 30% of all breast cancers. BRCA1 and BRCA2 are two major high-penetrance genes associated with hereditary breast and ovarian cancer syndrome, which together account for nearly half of inherited breast cancers. Other rare breast cancer susceptibility genes have been identified, such as CHEK2,TP53, PTEN, and STK11. Several emerging low-penetrance genes and loci recently discovered by genomewide association studies account for a small proportion of familial breast cancers (<5%). To date, about half of familial breast cancers remain unexplained but are likely attributable to as yet unknown genes and/or polygenic susceptibility. (From Olopade O, et al. Advances in breast cancer: pathways to personalized medicine. Clin Cancer Res 2008;14(24): Fig 1.)
The integral role of BRCA1 and BRCA2 in double-strand DNA repair holds potential as a therapeutic target for BRCA-related breast cancers. For example, platinum agents cause interstrand crosslinks, thereby blocking DNA replication and leading to stalled replication forks. Poly(adenosine diphosphate-ribose) polymerase-1 (PARP1) inhibitors additionally show promise as specific therapy for BRCA-related tumors. PARP1 is a cellular enzyme that functions in single-strand DNA repair through base excision and represents a major alternative DNA repair pathway in the cell.10,11 When PARP inhibition is applied to a background deficient in double-strand DNA repair, as is the case in BRCA-related tumor cells, the cells are left without adequate DNA repair mechanisms and ultimately undergo cell cycle arrest, chromosome instability, and cell death.4 Given their phenotypic similarities to BRCA1-related breast cancers, sporadic basal-like breast tumors may display sensitivity to PARP inhibition as well.11 Phase 2 studies are currently under way to explore the use of PARP inhibitors in both BRCA– and basallike, non-BRCA-related breast tumors.
Other High-Penetrance Genes
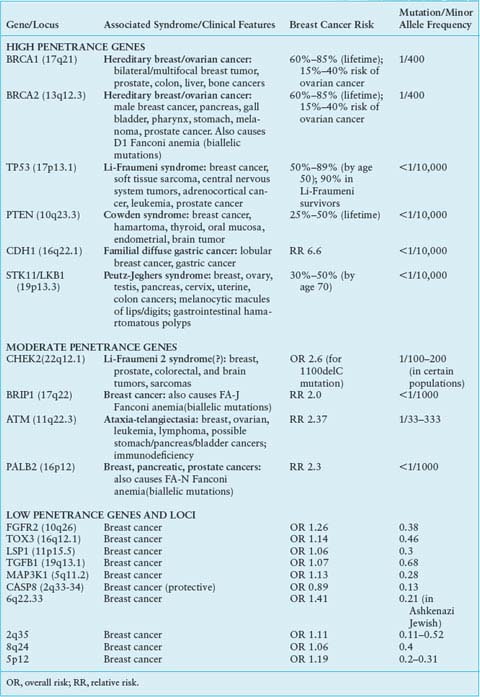
A small number of other high-risk, low frequency breast cancer susceptibility genes exist, and they include TP53, PTEN, STK11/LKB1, and CDH1.4–6 These high-penetrance genes confer an eight- to tenfold increase in risk of breast cancer as compared to noncarriers, but they collectively account for less than 1% of cases of breast cancer. Like BRCA1 and BRCA2, these genes are inherited in an autosomal dominant fashion and function as tumor suppressors.12 The hereditary cancer syndromes associated with each gene are usually characterized by multiple cancers in addition to breast cancer, as summarized in Table 24.1.
Moderate-Penetrance, Low Frequency Breast Cancer Predisposition Genes
Four genes have been identified that confer an elevated but moderate risk of developing breast cancer, namely CHEK2, ATM, BRIP1, and PALB2 (Table 24.1). Each of these genes confers approximately a two- to threefold relative risk of breast cancer in mutation carriers, though this risk may be higher in select clinical settings.5 Mutation frequencies in the general population are rare, on the order of 0.1% to 1%, though some founder mutations have been identified. Together, these genes account for approximately 2.3% of inherited breast cancer. The moderate relative risk of breast cancer of these genes in conjunction with the low population frequency renders this class of genes very difficult to detect with typical association studies. However, these genes were specifically selected for study as candidate breast cancer genes based on their known roles in signal transduction and DNA repair in close association with BRCA1 and BRCA2.6
TABLE 24.1
BREAST CANCER SUSCEPTIBILITY GENES AND LOCI
Low-Penetrance, High Frequency Breast Cancer Predisposition Genes and Loci
Both candidate gene and genome-wide association studies (GWAS) have identified a low-risk panel of approximately ten different alleles and loci in 15% to 40% of women with breast cancer5 (Table 24.1). Despite their frequency, the relative risk of breast cancer conferred by any one of these genetic variants alone is minimal, on the order of less than 1.5.4 Nevertheless, these alleles and loci may become clinically relevant in their suggestion of interactions with other high-, moderate-, and low-risk genes; these additive or multiplicative relationships could account for a measurable fraction of population risk. For example, association studies of FGFR2 and MAP3K1 within BRCA families showed that these single nucleotide polymorphisms (SNPs) conferred an increased risk in the presence of BRCA2 mutations.
Recent studies suggest that microRNA (miRNA) SNPs may also contribute to breast cancer susceptibility, and miRNAs appear to regulate many tumor suppressor genes (TSGs) and oncogenes via degradation of target mRNAs or repression of their translation. Thus, genetic variations in miRNA genes or miRNA binding sites could affect expression of TSGs or oncogenes and, thereby, cancer risk. For example, specific SNPs located within pre-miR-27a and miR-196a-2 genes have been associated with reduced breast cancer risk.13
SOMATIC CHANGES IN BREAST CANCER
The vast majority of breast cancers are sporadic in origin, ultimately caused by accumulation of numerous somatic genetic alterations.1 Recent data suggest that a typical individual breast cancer harbors anywhere from 50 to 80 different somatic mutations.2 Many of these mutations occur as a result of erroneous DNA replication; others may occur through exposure to exogenous and endogenous mutagens. To date, hundreds of candidate somatic breast cancer genes have been identified through GWAS.14,15 Yet the full range of somatic mutations will not be clear until hundreds more breast tumor samples are sequenced. To this end, international efforts are currently underway to produce a comprehensive catalog of these genetic alterations.
Determining the role of each identified mutation in the development of breast cancer remains a substantial challenge. Data suggest that the vast majority of identified somatic DNA mutations in a given tumor are “passenger” mutations, representing harmless, biologically neutral changes that do not contribute to oncogenesis.1,2 Conversely, “driver” mutations confer a growth advantage on the cell in which they occur and appear to be implicated in cancer development. By definition, driver mutations are found in candidate cancer genes (CAN).15
Although the catalog of somatic mutations and CAN genes is still incomplete, it is comprehensive enough that various structural features are starting to emerge. When specific driver mutations are cataloged among several different breast tumors, a bimodal cancer “genomic landscape” appears, comprising a small number of commonly mutated gene “mountains” among hundreds of infrequently mutated gene “hills.”1,2 Gene mountains correspond to the most frequently mutated genes found within breast tumors, such as TP53, CDH1, phosphatidylinositol 3-kinase (PI3K), cyclin D, PTEN, and AKT.6 Each individual gene hill, on the other hand, is typically found in less than 5% of breast tumors.1,16 This substantial heterogeneity of DNA mutations among breast tumors may explain the wide variations in phenotypes, both in terms of tumor behavior as well as responsiveness to therapy.
Historically, the focus of genetic research has been on the gene mountains, in part because they were the only mutations that available technology could identify. However emerging data suggest that it is actually the gene hills that play a much more pivotal role in breast cancer, consistent with the idea that having a large number of mutations, each associated with a small survival advantage, drives tumor progression. Recent studies have shown that a substantial number of these infrequent somatic mutations sort out among a much smaller number of biologic groups and cell signaling pathways that are known to be pathogenic in breast cancer, thereby vastly reducing the complexity of the genomic landscape. Examples of such pathways include interferon signaling, cell cycle checkpoint, BRCA1/2-related DNA repair, p53, AKT, transforming growth factor-β (TGF-β) signaling, Notch, epidermal growth factor receptor (EGFR), FGF, ERBB2, RAS, and PI3K. In short, it appears that common pathways, rather than individual gene mutations, govern the course of breast cancer development.16
Although recurrent point mutations are less common in breast cancer than other solid tumors, emerging data show that particular regions of the genome are commonly amplified and these regions contain genes that drive cancer progression (Table 24.2). The best example of an important amplified region is the 17q12 amplicon that harbors the HER2 oncogene. This amplicon leads to a more aggressive tumor phenotype, now the target of a highly successful antibody therapy, trastuzumab (Herceptin). It has been observed that RNAi knockdown of coamplified genes within the 17q12 amplicon results in decreased cell proliferation and increased apoptosis.17 Thus, the 17q12 amplicon appears to encode a concerted genetic program that contributes to the oncogenesis.
TABLE 24.2
RECURRENT AMPLIFICATIONS IN BREAST CANCER CELL LINES AND HUMAN TUMORS
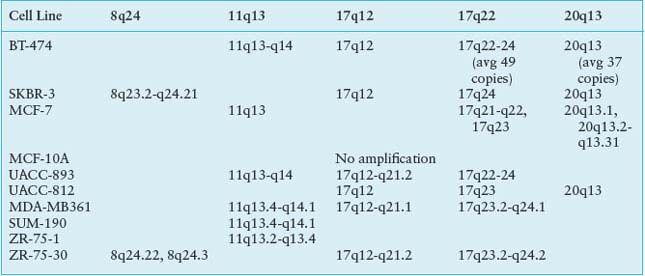
There are several other amplicons, in addition to 17q12 (HER2), that seem to drive the cancer phenotype and have prognostic significance in breast cancers, for example, 11q13 (CCDN1) and 8q24 (MYC), 20q13.18 These regions contain gene sets that are important in DNA metabolism and maintenance of chromosomal integrity, suggesting that response to DNA damaging agents used as anticancer therapy might be modulated by the presence of particular amplicons. Indeed, these coamplicons are frequent in HER2 amplified tumors and may modify tumor behavior and patient outcome.19,20 The contribution of these genomic alterations to functional consequences, may lie not in the overexpression of individual genes, but of gene cassettes on the amplicon.
Direct clinical translation of the growing catalog of somatic alterations in breast cancer has yet to evolve. However, with advancing technology and further identification and categorization of genetic mutations, new opportunities for individualized diagnosis and treatment options are likely to emerge.
GENE EXPRESSION PATTERNS IN BREAST CANCER
The cellular programs that are encoded by DNA are enacted by transcription into messenger RNA (mRNA) and translated into protein. Not surprisingly, the DNA alterations described above lead to either under- or overexpression of their associated mRNAs; consequently abnormal gene expression patterns are a common finding in breast tumors. Gene expression profiling has been introduced into the clinical literature during the past decade as research suggests that assessing the expression of multiple genes in a tumor sample may reflect programs turned on by DNA alternations and predict tumor behavior. So-called molecular signatures hold promise for improving diagnosis, prediction of recurrence, and selection of therapies for individual patients.
Several technologies have been developed to generate molecular signatures, including cDNA and oligonucleotide arrays and multiplex polymerase chain reaction (PCR) technologies. These technologies and newly developed statistical methodologies now allow evaluation of hundreds and even thousands of mRNAs simultaneously with grouping of samples based on coexpressed genes.
Molecular Classification of Breast Cancer
The seminal work by Perou et al.21 and Sorlie et al.22 suggests a classification of breast cancer subtypes based on gene expression patterns they termed “molecular portraits” of breast cancer. Among the categories they defined were the luminal A and B tumor types (typically estrogen receptor [ER] or progesterone receptor [PR] positive), HER2 gene-amplified tumors, and a newly recognized class termed “basal-like” due to the expression of basal keratins. Basal tumors typically lack ER, PR, and HER2, and are often referred to as triple negative, although not all basal-like tumors are triple negative and the reverse. Although the exact definition of molecular subtypes is an area of active debate, it is clear that these subtypes are reproducible in multiple, unrelated data sets, and their prognostic impact has been validated in these settings.21,23,24 As a result, clinical trials are now being designed to subdivide patients by ER/PR and HER2 status to validate claims that therapeutic approaches should address these groups rather than the population of breast cancer patients as a whole.
Prognostic Signatures
Around the same time period, van’t Veer et al.24 and van de Vijver et al.25 were the first to apply gene expression analysis to define a subgroup of breast cancer patients with increased likelihood of metastasis. The estimated hazard ratio for distant metastases in the group with a poor prognosis signature, as compared with the group with the good prognosis signature, was 5.1 (95% confidence interval, 2.9 to 9.0; P <.001). The European Organisation for Research and Treatment of Cancer (EORTC) and the Breast International Group (BIG) are currently conducting a prospective clinical trial to validate the utility of this assay for sparing patients from systemic chemotherapy (the MINDACT study).26 In a preliminary analysis, the 70-gene profile signature was strongly prognostic, outperforming classic prognostic criteria such as those used by the St. Gallen consensus panel27; however, the magnitude of effect was much less than previously reported with hazard ratios for time to distant metastases of 1.85 (1.14 to 3.0) and for overall survival of 2.5 (1.4 to 4.5). The 70-gene signature is now commercialized as the MammaPrint and has received clearance by the U.S. Food and Drug Administration (FDA) as a class 2, 510(k) product.
Other groups have developed prognostic gene expression signatures, including the 76-gene Rotterdam signature, which identifies a high-risk group of node-negative patients, and the Genomic Grade Index (GGI), which distinguishes poor and good prognosis groups in breast tumors of intermediate histologic grade.28 The potential value of these signatures has yet to be clearly defined, but it emphasizes the role of gene expression profiling at distinguishing prognostic groups not otherwise recognizable by standard histologic or clinical parameters.
Predictive Signatures
Endocrine Therapy
Several groups have applied gene expression profiling analysis to better define the likelihood of benefit from therapy. Such predictive signatures may have particular value as they help oncologists counsel patients about appropriate choices for treatment. Genomics Health Inc (Redwood City, California) developed the Oncotype DX® assay as a predictor of benefit from antiestrogen therapy using multiple real-time reverse transcriptase polymerase chain reaction (RT-PCR) assays in formalin fixed paraffin-embedded tissue. The assay was developed from 250 candidate genes selected from published literature, genomic databases, and in-house experiments performed on frozen tissue. From these data, a panel of 16 cancer-related genes and 5 reference genes were used to develop an algorithm to compute a recurrence score, ranging from 0 to 100, that can be used to estimate the odds of recurrence over 10 years from diagnosis.29 Paik et al.29 reported an analysis of two randomized controlled trials: National Surgical Adjuvant Breast and Bowel Project NSABP-B14, in which node-negative patients with ER-positive tumors were randomly assigned to tamoxifen or nil; and NSABP-B20, in which node-negative patients with ER-positive tumors were randomly assigned to tamoxifen alone or with cyclophosphamide, methotrexate, and fluorouracil (CMF) chemotherapy. Using the tissue samples from NSABP-B20, patients were categorized into three recurrence score groups: low risk (recurrence score less than 18), intermediate risk (recurrence score 18 to 30), and high risk (recurrence score 31 to 100). Samples from NSABP-B14 were then analyzed and found to be 6.8% (4.0% to 9.6%), 14.3% (8.3% to 20.3%) and 30.5% (23.6% to 37.4%). Paik et al. further analyzed the performance of the Oncotype DX assay to include patients in the other arms of NSABP-B14 and NSABP-B20 and found that the Oncotype DX assay was a strong predictor of benefit from CMF in NSABP-B20, with little or no benefit from chemotherapy for patients with low or intermediate recurrence scores but substantial benefit for those with high recurrence scores. Conversely, in NSABP-B14, the benefit from tamoxifen versus observation was confined to the low and intermediate risk categories (P value for interaction of .001). These data suggest that in patients who have an apparent favorable prognosis based on clinical features (negative nodes, positive ER), the Oncotype DX assay helps determine those most likely to benefit from tamoxifen only (low recurrence scores) versus those most likely not to benefit from tamoxifen but likely to benefit from chemotherapy (high recurrence scores). The benefits of chemotherapy in the 25% of patients who have intermediate recurrence scores remains uncertain and are the basis of an ongoing prospective randomized trial (Tailor Rx) where those with high recurrence scores will receive endocrine therapy and chemotherapy, those with low recurrence scores will receive endocrine therapy alone, and intermediate recurrence scores are randomly assigned to endocrine therapy versus endocrine and chemotherapy. A recent study by Albain et al.30 suggests that a low recurrence score predicts a lack of benefit of fluorouracil (5-FU), Adriamycin (doxorubicin), and cyclophosphamide (FAC) chemotherapy in node-positive breast cancer patients treated on Southwest Oncology Group SWOG-8814. Although these provocative data suggest a similar utility for Oncotype DX in node-positive patients, they do not include the use of taxanes and require additional validation with modern-day regimens. The value of the Oncotype DX assay in predicting benefit from hormonal therapy in patients treated with aromatase inhibitor therapy has recently been published, demonstrating that the assay performs equally similarly with both tamoxifen and anastrazole but does not distinguish benefit of one over the other.31
Additional predictors for ER-positive breast cancer include the Breast Cancer 2-Gene Expression Ratio (AvariaDx Inc, Carlsbad, California), a quantitative RT-PCR–based assay that measures the ratio of the HOXB6 and IL17BR genes, and is marketed as a marker of recurrence risk in untreated ER-positive/node-negative patients.32,33 The Breast Cancer Gene Expression Ratio is significantly and independently associated with poorer disease-free survival in two studies of lymph node–negative, ER-positive, tamoxifen-treated patients with breast cancer. In these two studies, patients who were low risk by the two-gene expression ratio had average 10-year recurrence rates of approximately 17% to 25%. Further validation is awaited.
Chemotherapy
Defining predictors of response to chemotherapy and targeted therapies has been more challenging. Ayers et al.34 from the M. D. Anderson Cancer Center were the first to report that multigene analysis of fine needle aspiration specimens predicts response to neoadjuvant T-FAC chemotherapy. These results require validation and are the subject of a prospective trial of chemotherapy with randomization based on the predictor (F. Symmans, personal communication). Another approach to the development of predictive signatures was pioneered by the group at Duke Center for Health Policy and Informatics where gene signatures were defined in model systems and applied to human data sets. While initially tested in lung cancer, these signatures are now the subject of several prospective randomized trials in breast cancer, and other tumor types.35 Validation of gene signatures is of utmost importance in the future to determine the value of these expression profiles at predicting treatment response, and clinical outcome, in breast cancer patients. National organizations such as the American Society of Clinical Oncology, National Comprehensive Cancer Network, and College of American Pathologists have ongoing efforts to interpret the data from the burgeoning field of multigene biomarker tests to help the practicing clinician interpret their clinical utility.36
EPIGENETICS OF BREAST CANCER
Cells maintain their stable identity and phenotype over many generations without external stimuli or signaling events. This cellular memory is encoded in the epigenome, a collection of heritable information that exists alongside the genomic sequence. DNA methylation and chromatin modification are major epigenetic mechanisms in higher eukaryotes and are tightly coupled to basic genetic processes, such as DNA replication, transcription, and repair. DNA methylation at the promoter proximal CpG sequences is associated with gene silencing. Similarly, specific histone modifications control transcriptional status or capacity of the underlying sequence by regulating the activity of chromatin domains in an active or inactive state. Euchromatin or active chromatin is enriched with acetylated histones H3, H4, and H2A and histone H3 methylated at lysine residues K4, K40, and K36.37 In contrast, heterochromatin or silent chromatin is depleted of histone acetylation while enriched in histone H3 methylated at lysine resides K9, K27, and K79. It is well documented that cancers, including breast cancer, have altered patterns of DNA methylation and histone acetylation, leading to alterations in transcription that appear to be oncogenic.37,38 Hence, epigenetic therapies have received intense interest from a large number of clinical and basic cancer studies.
Major epigenetic cancer drugs include DNA methyltransferase (DNMT) and histone deacetylase (HDAC) inhibitors. Preclinical studies show promise that HDAC inhibitors may have activity in breast cancer cells, and many clinical phase 1 and 2 studies are in progress.39,40
MicroRNAs: A Newly Discovered Class of Molecules that Regulate Gene Expression
miRNAs are small noncoding RNAs that belong to a novel class of regulatory molecules that control expression of hundreds of target mRNA transcripts. miRNAs are generated from large RNA precursors (termed pre-miRNAs) that are processed in the nucleus into approximately 70 nucleotide pre-miRNAs, which fold into imperfect stem-loop structures.41 The pre-miRNAs undergo an additional processing step within the cytoplasm, and mature miRNAs of 18 to 25 nucleotides in length are excised from one side of the pre-miRNA hairpin by Dicer. miRNAs regulate gene expression in two ways. First, miRNAs that bind to protein-coding mRNA sequences that are exactly complementary to the miRNA induce the RNA-mediated interference (RNAi) pathway. Messenger RNA targets are then cleaved by ribonucleases in the RNA-induced silencing complex (RISC). Second, miRNAs bind to imperfect complementary sites within the 3’-untranslated regions (3’UTRs) of their target protein-coding mRNAs and repress the expression of these genes at the level of translation.42
miRNAs are known to be associated with breast cancer in both cell lines and clinical samples. For example, miR-21, miR-155, miR-7, and miR-210 are overexpressed in aggressive human breast cancers,43,44 while let-7 and miR-125a have been shown to be down-regulated in breast cancers.45 It has also been shown that miR-125a may function as a tumor suppressor by inhibiting ERBB2 and ERBB3.
MicroRNAs Predict Response to Cancer Treatment
Soon after they were discovered to be misregulated in cancer, miRNA misexpression patterns were found to be associated with cancer outcome and response to treatment, including radiation and chemotherapy. Certain miRNAs associated with hypoxia, such as miR-210, have been shown to be biomarkers of poor outcome in breast cancer.43 Furthermore, in vitro data show that certain miRNAs are associated with resistance to doxorubicin46 or tamoxifen.47 In patient samples, an association of miRNA’s tumor subtypes have specific miRNA patterns and this is associated with poor outcome. Defining the role of miRNAs as biomarkers for prognosis and prediction, as well as their potential as targeted therapies, is an active area of research in breast cancer.
PROTEIN/PATHWAY ALTERATIONS
The molecular mechanisms that lead to cancer have been characterized as the hallmarks of cancer, as proposed by Hanahan and Weinberg.48 They include self-sufficiency in growth signals, insensitivity to antigrowth factor signals, evasion of apoptosis, infinite replicative potential, invasion and metastasis, and sustained angiogenesis. The effectors of genetic and epigenetic abnormalities are in most cases reflected in the abnormal levels, functions, and interactions of proteins and signaling pathways. Undoubtedly, numerous alterations coordinate to result in the malignant phenotype; however, a number of key proteins and their pathways have emerged as critical drivers of breast cancer development and growth as well as potential therapeutic targets.
Therapeutic Targets in Breast Cancer
Estrogen Signaling
Most breast cancers are intimately linked with exposure to estrogen and alterations in the estrogen receptor signaling pathway. Estrogen is a steroid hormone that exerts its actions by binding to the nuclear ER. Upon activation by its ligand, ER binds in a coordinated fashion with a number of coregulatory proteins to estrogen response elements in the promoter region of estrogen-responsive genes. This in turn directs the transcription of numerous growth-promoting genes, including PR. Although ER is overexpressed in as many as 70% of invasive breast cancers, the precise mechanism by which this occurs is unclear. Amplification of the gene appears to be one mechanism; however, it was present in only approximately 50% of cases with ER overexpression in one study, suggesting that transcriptional deregulation and posttranscriptional modifications (such as alteration of mRNA levels by miRNAs) may also play a role. The level of ER expression is not only of biologic interest, but it is a highly effective predictor for response to antiestrogens, which is a recommended treatment for all ER-expressing tumors.
Estrogen exerts its actions through both genomic (described above) and nongenomic mechanisms. In contrast to the genomic actions of ER, nongenomic actions of ER are extremely rapid (within seconds to minutes of estrogen exposure) and are believed to result from the hormone-dependent activation of membrane-bound or cytosolic ERs. These nonnuclear ER actions result in rapid phosphorylation and activation of important growth regulatory kinases including EGFRs, insulinlike growth factor-1R (IGF-1R), c-Src, Shc, and the p85α regulatory subunit of PI3K.5 This “crosstalk” between ER and growth factor receptors is bidirectional: constitutive HER2, for example, can increase ER signaling to the point where it is unresponsive to antiestrogen treatments. These findings suggest a role for HER2/IGF-1R/EGFR activation in both acquired and de novo resistance to treatment with antiestrogens.49
The ER pathway has proven to be an invaluable target for therapeutic treatments in breast cancer. A number of agents have been developed over the prior decades that can inhibit this pathway by either binding to the receptor itself (e.g., selective ER modulators such as tamoxifen) or by decreasing the production of endogenous estrogen (e.g., aromatase inhibitors and ovarian ablation). Although these agents are highly effective and have made a significant impact on breast cancer morbidity and mortality, unfortunately, de novo and acquired resistance is also quite common.
As described above, the Oncotype DX assay adds additional insight into the behavior of ER-positive tumors and provides useful information for treatment decision making.
Growth Factor Receptor Pathways
Growth factor receptor pathways, and in particular tyrosine kinase receptors, play an essential role in initiating both proliferative and cell survival pathways in tissue and are normally tightly regulated. In breast cancer biology, the ErbB family has been studied most extensively, but an expanding number of other growth factors, such as insulinlike growth factor receptors, have also been the subject of intense scrutiny in hopes of identifying effective therapeutic targets.50 These receptors have an extracellular ligand-binding region, a transmembrane region, and a cytoplasmic tyrosine kinase–containing domain that can activate downstream signaling cascades. These growth factor receptor pathways can be constitutively activated by a number of mechanisms, including excessive ligand levels, gain-of-function mutations, overexpression with or without gene amplification, and gene rearrangements and resultant fusion proteins with oncogenic potential. This can ultimately lead to inappropriate kinase activity and growth promoting second messenger activation (Fig. 24.2).
Human Epidermal Growth Factor Receptor 2
HER2 (EGFR2 or ErbB2) is a member of a family of receptor tyrosine kinases that also includes EGFR (HER1, ErbB1), ErbB3, and ErbB4. Ligand binding to the extracellular domains of the ErbB1, ErbB3, or ErbB4 receptors induces homo- and heterodimerization and kinase activation. The HER2 protein exists in a closed conformation and has no ligand, but it is the preferred partner for dimerization with HER1, -3, and -4. At a molecular level, HER2 amplification is associated with deregulation of G1/S phase cell cycle control via up-regulation of cyclins D1, E, and cdk6, as well as p27 degradation. HER2 also interacts with important second messengers including SH2 domain-containing proteins (e.g., Src kinases).
More important, HER2 amplification or protein overexpression (found in 20% to 30% of invasive breast cancers) is clearly associated with accelerated cell growth and proliferation, poor clinical outcome, and response to the monoclonal anti-HER2 antibody, trastuzumab. In the adjuvant setting, several independent randomized studies have shown that the addition of trastuzumab to chemotherapy reduced the rate of recurrence by over 50% among women with HER2-positive early stage breast cancer.51 Based on these results, trastuzumab in combination with standard chemotherapy was approved by the FDA in 2006 for use in the adjuvant setting.
The precise mechanism of action of trastuzumab (and therefore mechanism of resistance as well) remains controversial. Trastuzumab appears to inhibit several major pathways that regulate tumor growth. First, trastuzumab may disrupt heterodimeric interaction of HER2 (and also block cleavage of HER2 at the juxtamembrane region). Second, trastuzumab modulates host immunity, activating natural killer cells involved in antibody-dependent cellular cytotoxicity (ADCC). In both animal models and more recently in clinical studies, the FcγR genotype required for ADCC was associated with trastuzumab effectiveness in HER2 overexpressed breast cancer. Third, trastuzumab also appears to affect angiogenesis at multiple levels, including a decrease in tumor-associated microvessel density, reduction in proangiogenic factors, and “normalization” of neovascularization. Finally, trastuzumab partially inhibits numerous downstream signaling pathways, most notably the ras/raf/MEK/MAPK and the PI3K/AKT pathways, and studies suggest that activation of these pathways by alternate receptors (e.g., IGF-1R, c-met) or deregulation through loss of PTEN or P13K mutation may lead to resistance to trastuzumab52 (Fig. 24.2).
Human Epidermal Growth Factor Receptor 2/Epidermal Growth Factor Receptor
As HER2 signaling is effected through heterodimer formation with other EGFRs, targeting HER2 and EGFR simultaneously, for example, may provide therapeutic synergy. The tyrosine kinase inhibitor lapatinib competes with adenosine triphosphate to bind to the activation loop of target kinases, thereby inhibiting their activity. Lapatinib inhibits the tyrosine phosphorylation of both EGFR and HER2 and in turn inhibits activation of the proproliferative kinases ERK1/2 and AKT. Lapatinib has now been FDA approved in combination with capecitabine in advanced HER2-positive breast cancer previously progressed on trastuzumab, based on a phase 3 trial showing a significant benefit for the combination over capecitabine alone.
Insulinlike Growth Factor-1R
In addition to activation of the EGFR pathway, signaling via insulinlike growth factor (IGF-1 and IGF-2) and their receptor (IGF-1R) can result in phosphorylation and activation of a variety of oncogenic kinases. IGF-1R is the primary response mediator of IGF and is expressed in all epithelial cell types. However, elevated IGF-1 levels and IGF-1R signaling have been implicated in increased risk of breast and other cancers. The IGF-1R has in particular been shown to be an effective target in preclinical studies. Similar to ErbB receptor inhibition, IGF-1R inhibitors have focused primarily on neutralizing antibodies and tyrosine kinase inhibitors. Currently more than eight monoclonal antibodies directed against IGF-1R are in various stages of clinical development in a variety of cancers. Notably, extensive cross-talk of IGF-1R downstream signaling pathways with other important signaling pathways in breast cancer suggests that combining IGF-1R inhibitors with other targeted agents may be an effective therapeutic strategy. For example, IGF-1R up-regulation has been suggested as a mechanism of resistance to HER2 and EGFR inhibitors through sustained activation of the PI3K/AKT pathway. Similarly, IGF-1R up-regulation has also been seen in breast cancer cells that have developed resistance to antiestrogen and HER2 treatments.
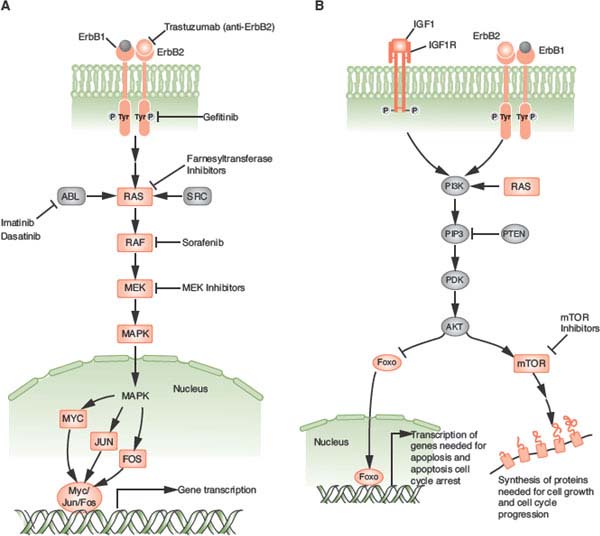
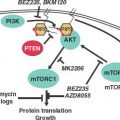
FIGURE 24.2 A: The ras/raf/MEK/MAPK pathway is activated by multiple growth factor receptors (here exemplified by ErbB1 and ErbB2) as well as several intracellular tyrosine kinases such as SRC and ABL. Activated RAS stimulates a sequence of phosphorylation events mediated by RAF, MEK, and ERK (MAP) kinases. Activated MAP kinase (MAPK) translocates to the nucleus and activates proteins such as MYC, JUN, and FOS that promote the transcription of numerous genes involved in tumor growth. B: The phosphatidylinositol 3-kinase (PI3K)pathway is activated by RAS and by a number of growth factor receptors (here exemplified IGF1R and the ErbB1/ErbB2 heterodimer). Activated PI3K generates phosphatidylinositol-3,4,5-triphosphate (PIP3), which activates phosphoinositide-dependent kinase-1 (PDK). In turn, PDK phosphorylates AKT. PTEN is an endogenous inhibitor of AKT activation. Phosphorylated AKT transduces multiple downstream signals, including activation of the mammalian target of rapamycin (mTOR) and inhibition of the FOXO family of transcription factors. mTOR activation promotes the synthesis of proteins required for cell growth and cell cycle progression. (Redrawn from Golan DE, Tashjian AH, Armstrong EJ. Principles of pharmacology: the pathophysiologic basis of drug therapy. 2nd ed. Baltimore: Wolters Kluwer Health, 2008.)
Ras and Phosphatidylinositol 3-Kinase Pathways
Redundancies and cross-talk of numerous different signaling pathways are a common theme. Several downstream messengers, however, bear special consideration due to their functional importance and therapeutic implications. PI3K/AKT is a central signaling pathway downstream of many receptor tyrosine kinases and regulates cell growth and proliferation (Fig. 24.2B). Activating mutations in the gene encoding the p110α catalytic subunit of PI3K (PI3CKA) may be an important contributing factor to mammary tumor progression. Activating mutations of the AKT gene family are rare.
PTEN dephosphorylates, and therefore inactivates, the p110 catalytic domain of PI3K and is either mutated or underexpressed (e.g., via methylation) in many breast cancers. Activation of the PI3K pathway, in turn, results in the 3-phosphoinositide-dependent kinase-mediated activation of several known kinases including AKT1, AKT2, and AKT3. Interestingly, activated AKT1 appears to be antiapoptotic but also plays an antiinvasive role in tumor formation. In addition to the AKTs, downstream proliferative effectors of the PI3K pathway also include the mammalian target of rapamycin (mTOR) complex 1 (TORC1), which consists of mTOR, raptor, and mLst8. It is currently believed that TORC1 mediates its progrowth effects through the activation of S6-kinase1 and suppression of 4E-BP1, an inhibitor of cap-dependent translation. These observations all point to mTOR-raptor as a critical target in cancer therapy, and indeed, several mTOR inhibitors known as rapamycin analogues (e.g., CCI-779, RAD-001, AP-23576) are undergoing clinical trials for the treatment of breast cancer.
The ras/raf/MEK/MAPK pathway is also a critical signaling pathway for numerous growth factor receptors (Fig. 24.2A). Thus far in breast cancer, agents that target the MEK pathway (e.g., raf inhibitor sorafenib) have had modest success as single agents, but studies in combination with other treatments hold more promise.
Angiogenesis
Angiogenesis is normally a tightly regulated process of vessel formation during physiologic events such as wound healing and pregnancy. It has also been shown to be an important part of tumor growth and spread. In contrast to physiologic angiogenesis, tumor-associated angiogenesis is highly dysregulated with disorganized and distorted vasculature and increased vascular permeability. Thus, in recent years, angiogenesis has become a frequent target for the treatment of many cancers.
Central to this process is the proangiogenic factor, vascular endothelial growth factor (VEGF), which together with its receptors, regulate endothelial cell growth and new vessel formation.53 VEGFRs, like EGFRs, are also tyrosine kinase receptors. VEGF-A binds to both VEGFR1 (Flt-1) and VEGFR2 (KDR/Flk1). VEGFR2 appears to mediate most of the known cellular responses to VEGFs, while the function of VEGFR1 is less well defined. Bevacizumab, a humanized monoclonal antibody directed against VEGF-A, has been the most extensively studied thus far. To date, three large randomized trials have shown a statistically significant benefit in progression-free survival when bevacizumab was added to a variety of different chemotherapies in the first-line metastatic setting. Multitargeted VEGFR tyrosine kinase inhibitors such as sunitinib (VEGFR, PDGFR, and c-kit blockade) and sorafenib (VEGFR and RAF kinase blockade) have also been studied extensively in breast and other cancers. Despite the success of some of these agents, identification of predictive factors for antiangiogenic response have thus far proven to be elusive and is not recommended as a selection criteria for treatment or for entry into clinical trials.
Selected References
The full list of references for this chapter appears in the online version.
1. Bell DW. Our changing view of the genomic landscape of cancer. J Pathol 2010;220(2):231.
2. Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007;318(5853):1108.
4. Turnbull C, Rahman N. Genetic predisposition to breast cancer: past, present, and future. Annu Rev Genomics Hum Genet 2008;9:321.
5. Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 2008;359(20):2143.
6. Hirshfield KM, Rebbeck TR, Levine AJ. Germline mutations and polymorphisms in the origins of cancers in women. J Oncol 2010;2010:297671.
7. Narod SA. Modifiers of risk of hereditary breast cancer. Oncogene 2006;25(43):5832.
8. Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene 2006;25(43):5846.
9. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002;108(2):171.
10. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009;361(2):123.
11. Iglehart JD, Silver DP. Synthetic lethality—a new direction in cancer-drug development. N Engl J Med 2009;361(2):189.
12. Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat Genet 2008;40(1):17.
13. Hoffman AE, Zheng T, Yi C, et al. microRNA miR-196a-2 and breast cancer: a genetic and epigenetic association study and functional analysis. Cancer Res 2009;69(14):5970.
14. Forbes SA, Bhamra G, Bamford S, et al. The catalogue of somatic mutations in cancer (COSMIC). Curr Protoc Hum Genet 2008;chapter 10:unit 10.11.
15. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009;458(7239):719.
16. Copeland NG, Jenkins NA. Deciphering the genetic landscape of cancer—from genes to pathways. Trends Genet 2009;25(10):455.
17. Kao J, Pollack JR. RNA interference-based functional dissection of the 17q12 amplicon in breast cancer reveals contribution of coamplified genes. Genes Chromosomes Cancer 2006;45(8):761.
18. Chin K, DeVries S, Fridlyand J, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006;10(6):529.
19. Bentires-Alj M, Gil SG, Chan R, et al. A role for the scaffolding adapter GAB2 in breast cancer. Nature Med 2005;12(1):114.
21. Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406(6797):747.
22. Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001;98(19):10869.
23. Gruvberger S, Ringnér M, Chen Y, et al. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res 2001;61(16):5979.
24. van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415(6871):530.
25. van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347(25):1999.
26. Piccart MJ, Loi S, Van’tVeer L, et al. Multi-center external validation study of the Amsterdam 70-gene prognostic signature in node negative untreated breast cancer: are the results still outperfoming the clinical-pathological criteria? Breast Cancer Res Treat 2004;88(S17): (abst 38).
27. Goldhirsch A, Wood WC, Gelber RD, et al. Meeting highlights: updated international expert consensus on the primary therapy of early breast cancer. J Clin Oncol 2003;21(17):3357.
28. Sotiriou C, Wirapati P, Loi S, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst 2006;98(4):262.
29. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351(27):2817.
30. Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010;11(1):55.
31. Mamounas E, Tang G, Fisher B, et al. Association between the 21-gene recurrence score assay and risk of locoregional recurrence in node-negative, estrogen receptor-positive breast cancer: results from NSABP B-14 and NSABP B-20. J Clin Oncol 2010;28(10):1677.
32. Ma XJ, Hilsenbeck SG, Wang W, et al. The HOXB13:IL17BR expression index is a prognostic factor in early-stage breast cancer. J Clin Oncol 2006;24(28):4611.
33. Ma XJ, Wang Z, Ryan PD, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell 2004;5(6):607.
34. Ayers M, Symmans WF, Stec J, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol 2004;22(12):2284.
35. Acharya CR, Hsu DS, Anders CK, et al. Gene expression signatures, clinicopathological features, and individualized therapy in breast cancer. JAMA 2008;299(13):1574.
36. Harris L, Fritsche H, Mennel R, et al. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol 2007;25(33):5287.
37. Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007;129(4):823.
38. Veeck J, Esteller M. Breast cancer epigenetics: from DNA methylation to microRNAs. J Mammary Gland Biol Neoplasia 2010;15(1):5.
39. Fiskus W, Ren Y, Mohapatra A, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res 2007;13(16):4882.
40. Zhou Q, Shaw PG, Davidson NE. Inhibition of histone deacetylase suppresses EGF signaling pathways by destabilizing EGFR mRNA in ER-negative human breast cancer cells. Breast Cancer Res Treat 2009;117(2):443.
42. Reinhart B, Slack FJ, Basson M, et al. The 21 nucleotide let-7 RNA regulates C. elegans developmental timing. Nature 2000;403:901.
43. Camps C, Buffa FM, Colella S, et al. hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res 2008;14(5):1340.
44. Foekens JA, Sieuwerts AM, Smid M, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci U S A 2008;105(35):13021.
45. Iorio MV, Casalini P, Tagliabue E, et al. microRNA profiling as a tool to understand prognosis, therapy response and resistance in breast cancer. Eur J Cancer 2008;44(18):2753.
48. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100(1):57.
50. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2(2):127.
51. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med 2007;357(1):39.
52. Hynes NE, Dey JH. PI3K inhibition overcomes trastuzumab resistance: blockade of ErbB2/ErbB3 is not always enough. Cancer Cell 2009;15(5):353.
53. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 2008;8(8):579.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


