Cancer is caused by a series of events that include the accumulation of successive molecular lesions and alterations in the tumor microenvironment (1). Molecular lesions include overexpression, amplification, or mutations of oncogenes; deletion of tumor suppressor genes; and the inappropriate expression of growth factors and their cellular receptors. In addition to these molecular changes, the formation of new blood vessels (angiogenesis) and the lack of effective host antitumor immune responses create a microenvironment that supports the growth of cancer (2). Our improved understanding of these mechanisms presents an opportunity for the development of novel therapeutic approaches (3). This chapter provides an overview of biologic, targeted, and immunotherapeutic strategies for gynecologic cancers.
Biologic and Targeted Therapies
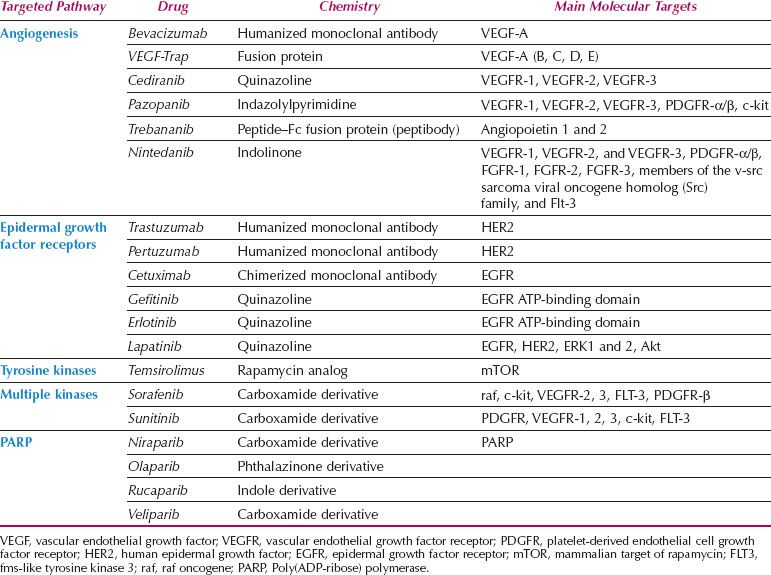
The growth of cancer cells is crucially dependent on oncogenic signal transduction pathways. Extracellular signals are transmitted to the cancer cell via transmembrane receptors. Activation of the epidermal growth factor receptors (EGFRs, HER2, HER3, and HER4), for example, stimulates a cascade of intracellular proteins that ultimately lead to changes in gene expression. Novel therapeutics are targeted to modulate these signal transduction pathways by blocking the extracellular transmembrane receptors or interfering with intracellular proteins such as tyrosine kinases further downstream. This novel therapeutic approach is also termed molecular targeting (4). It is accomplished by either monoclonal antibodies that bind to transmembrane receptors and serum proteins such as vascular endothelial growth factor (VEGF) or chemical, small-molecule inhibitors that prevent activation of signal transduction proteins. Targeting the signaling cascade inhibits the proliferation of cancer cells, induces apoptosis, and blocks metastasis. The specificity of these molecules is based on the assumption that cancer cells are overexpressing various proteins in the signal transduction pathways, therefore presenting a preferred target compared to normal cells. Conceptually, this should result in more cancer cell–specific therapy and less clinical side effects because of sparing of normal tissue (5). At this time, a large variety of molecular-targeting strategies are being tested for efficacy in clinical trials (Table 2.1).
Table 2.1 Targeted Cancer Therapies
Angiogenesis
The formation of new blood vessels (neoangiogenesis) is a normal process during embryonic development, tissue remodeling, and wound healing (6). Malignant tumors are able to induce angiogenesis by secreting paracrine factors that promote the formation of new blood vessels. Angiogenesis is a complex process that is influenced by various pro- and antiangiogenic factors, including VEGF, interleukin 8, platelet-derived endothelial cell growth factor, and angiopoietins. Overexpression of these angiogenic factors leads to neovascularization and increased supply of nutrients and oxygen to the tumor.
Three main therapeutic strategies that target angiogenesis are currently being explored for the treatment of cancer patients (7). One group of agents targets VEGF (e.g., bevacizumab, VEGF-Trap), the second group prevents VEGF from binding to its receptor (pertuzumab), and the third group of agents inhibits tyrosine kinase activation and downstream signaling in the angiogenesis signaling cascade (vatalanib, sunitinib) (8).
Vascular Endothelial Growth Factor
VEGF is overexpressed in gynecologic malignancies, therefore presenting an excellent target for therapy (9). Inhibition of VEGF-induced angiogenic signaling decreases tumor microvascular density and causes death of solid tumors in various preclinical models. Several agents are now available for clinical use; all target the VEGF signaling pathway. The most widely used agent at this time is bevacizumab, a humanized, recombinant monoclonal antibody that binds to all isoforms of VEGF-A (12). Bevacizumab has been approved for the treatment of colorectal carcinoma based on improved overall survival in combination with chemotherapy in patients with metastatic disease, single agent therapy in glioblastoma with progressive disease, first-line treatment in combination with carboplatin and paclitaxel for lung cancer, and in combination with interferon alpha for metastatic renal cell carcinoma (10,11).
In ovarian carcinoma, various clinical trials have demonstrated the efficacy of bevacizumab treatment (see Chapter 11 for more discussion). The initial studies by the Gynecologic Oncology Group included 62 patients treated with single agent bevacizumab 15 mg/kg intravenously every 21 days (13). Thirteen patients (21%) showed clinical responses with two complete and eleven partial responses. The median response duration was 10 months, and 25 patients (41.3%) survived progression free for at least 6 months. In a second trial, bevacizumab treatment of 44 patients with recurrent, platinum-resistant ovarian carcinoma resulted in partial responses in seven patients (15.9%) and stable disease in 27 (61.4%) (14). Median progression-free survival (PFS) was 4.4 months with a median survival of 10.7 months.
Based on these promising data, Phase III first-line clinical trials of bevacizumab were conducted. The GOG-0218 and ICON7 studies assessed the efficacy of bevacizumab added to carboplatin and paclitaxel followed by maintenance therapy in patients with epithelial ovarian cancer (15,16). Significant improvements were demonstrated in progression-free survival in both studies with concurrent and maintenance bevacizumab treatment. In addition, an overall survival benefit of almost 8 months (28.8 vs. 36.6 months; HR, 0.64; 95% confidence interval [CI], 0.48 to 0.85; p < 0.002) was found for the subgroup of patients with high-risk disease when treated with bevacizumab. This high-risk group was defined as having FIGO stage IV or stage III disease with suboptimal tumor debulking. Bevacizumab did not show an overall survival benefit when the entire cohort was analyzed.
Bevacizumab has likewise demonstrated efficacy in recurrent ovarian cancer, both in the so-called AURELIA and OCEANS studies (17,18). In the AURELIA trial, patients with platinum-resistant disease were treated with bevacizumab in combination with chemotherapy (either topotecan, pegylated liposomal doxorubicin, or weekly paclitaxel). A statistically significant improvement in PFS (3.4 vs. 6.7 months; HR, 0.48; p < 0.001) was found in patients treated with the combination. In the OCEANS study, bevacizumab in combination with carboplatin and gemcitabine followed by maintenance therapy in platinum-sensitive patients resulted in a significant improvement in PFS compared to chemotherapy alone (8.4 vs. 12.4 months; HR, 0.48; p < 0.0001). Neither the OCEANS nor AURELIA trial showed a benefit in overall survival, but both demonstrated the efficacy of bevacizumab even in platinum-resistant disease.
A recent trial conducted by the GOG has demonstrated the efficacy of bevacizumab in combination with cisplatin and paclitaxel or cisplatin with topotecan in locally advanced and metastatic cervical cancer. The median PFS in the bevacizumab group was 8.2 months, compared with 5.9 months in the chemotherapy alone group. In addition, the median overall survival (OS) for patients who received cisplatin plus paclitaxel was 14.3 months, significantly less than the 17.5 months for the bevacizumab group (p = 0.0348). Similarly, the median OS for those who received topotecan plus paclitaxel was 12.7 months, compared with 16.2 months with bevacizumab (p = 0.0896).
Bevacizumab–related side effects include venous and arterial thrombosis, hemorrhage, nephrotic syndrome with proteinuria, hypertension, rare leukoencephalopathy, and bowel perforation (20–24). There are no predictors of response to bevacizumab at this time, but prospective clinical trials are currently investigating the utility of novel imaging technologies to monitor the clinical response.
Other antiangiogenic strategies are being pursued in clinical trials. Angiopoietin 1 and 2 have been identified as important mediators of angiogenesis in ovarian cancer via their binding to the Tie2 receptor. Trebananib (AMG386) is a peptide–Fc fusion protein that blocks the interactions between angiopoetin-1 and angiopoetin-2, which are expressed on vascular endothelial cells with the Tie2 receptor (19). This results in blocking of VEGF stimulation and vascular maturation. The ongoing first-line (TRINOVA-3) and recurrent (TRINOVA-1 and 2) ovarian cancer clinical trials combine trebananib with chemotherapy, and also use it as maintenance.
VEGF-Trap (AVE 0005) is a recombinant fusion protein that consists of the extracellular domain of VEGF receptors VEGFR1 and VEGFR2 fused to the FC portion of immunoglobulin G1 (25). VEGF is inactivated by binding to the ligand-binding domain of this fusion protein followed by destruction of this complex via immune system–mediated mechanisms. Small-molecule tyrosine kinase inhibitors that target the VEGF pathway include pazopanib, vatalanib, and sunitinib. Pazopanib is a multitarget tyrosine kinase inhibitor which is directed against VEGFR1, VEGFR2, VEGFR3, platelet-derived growth factor receptor (PDGFR), and c-kit (3). Similarly, vatalanib (PTK787) targets multiple VEGF-receptor tyrosine kinases. Sunitinib (SU11248) inhibits PDGR, VEGFR, c-kit, and SLT3, and has shown promising results in renal cell cancer and gastrointestinal stromal tumors. Trials in gynecologic malignancies are ongoing.
Epidermal Growth Factor Receptor
The epidermal growth factor receptor pathway plays an important role in regulation of growth and differentiation of epithelial cells through regulation of cell division, migration, adhesion, differentiation, and apoptosis (26). The epidermal growth factor receptor family consists of four members including EGFR (HER1), HER2, HER3, and HER4 (27). EGFR overexpression has been reported in 35–70% of patients with epithelial ovarian cancer (28,29). In endometrial cancer, EGFR is overexpressed in 43–67% of tumors, and is associated with a shortened disease-free and overall survival (30–32). In addition, amplification of the HER2 gene is commonly found in endometrial carcinoma. Overexpression of the HER2 receptor is more prevalent in nonendometrial cancer and is associated with an aggressive form of the disease. In uterine serous carcinomas, HER2 gene amplification can be demonstrated in as many as 42% of cases (33).
Various agents directed against epidermal growth factor receptors are available (34). Trastuzumab is a humanized monoclonal antibody that binds to the extracellular domain of HER2 (35). Blockade of HER2 affects various molecules that ultimately decrease cell proliferation. Pertuzumab is another humanized monoclonal antibody that binds to a different epitope of HER2 compared to trastuzumab. Binding to HER2 prevents dimerization of the receptor, which is required for its function (36). Cetuximab is a chimeric monoclonal antibody that binds to EGFR, thereby preventing dimerization and activation (37). Gefitinib is a small-molecule tyrosine kinase inhibitor of EGFR that prevents phosphorylation of the receptor by binding to the intracellular ATP-binding domain of the receptor (38). Erlotinib is a small-molecule tyrosine kinase inhibitor of EGRF that prevents phosphorylation of the intracellular domain of the EGFR receptor. Lapatinib (GW572016) inhibits both EGFR and HER2 (Fig. 2.1).
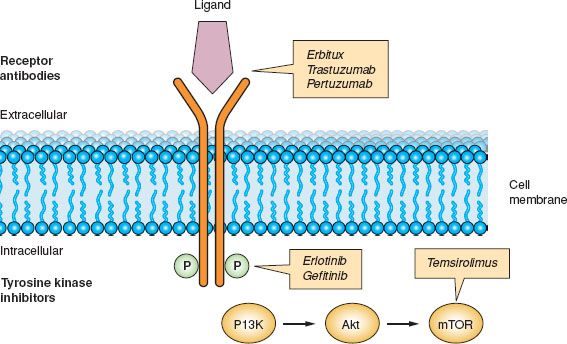
Figure 2.1 Inhibition of epidermal growth factor receptor signaling.
Inhibition of EGFR signaling is accomplished by using either monoclonal antibodies against the extracellular receptor or small-molecule inhibitors against the intracellular kinase domain. Both strategies result in inhibition of phosphorylation or receptor activation.
Erlotinib is a potent reversible inhibitor of EGFR tyrosine kinase that blocks receptor autophosphorylation and has been used for the treatment of ovarian carcinoma. In one study, 34 patients were treated with single agent erlotinib (150 mg/day orally) for as long as 48 weeks (39). Two patients showed a partial response lasting 8 and 17 weeks. Fifteen patients (44%) had stable disease, and 17 patients (50%) progressed under treatment. The side effects of erlotinib were mainly confined to tissues with strong expression of EGFR: skin rashes and diarrhea were observed in 68% and 38% of patients, respectively.
Erlotinib has been used in combination with docetaxel and carboplatin as first-line treatment after surgical cytoreduction in patients with ovarian, fallopian tube, and peritoneal cancers (40). In this study, 23 evaluable patients showed five complete and seven partial responses. The treatment was well tolerated; main side effects were neutropenia and skin rashes. The study demonstrated the feasibility and tolerability of erlotinib in conjunction with chemotherapy.
Cetuximab (C225, Erbitux) is a chimerized monoclonal antibody against EGFR. Treatment of patients with primary ovarian or peritoneal cancer using cetuximab has shown only modest activity in screened patients with EGFR-positive tumors. Cetuximab in combination with carboplatin resulted in three complete (10.7%) and six partial (21.4%) responses in 28 patients with recurrent ovarian cancer (41). Twenty-six of these 28 patients (92.8%) had EGFR-positive tumors. The combination of paclitaxel, carboplatin, and cetuximab for first-line chemotherapy of stage III ovarian cancer patients resulted in progression-free survival of 14.4 months and was therefore not significantly prolonged compared to historical data (42).
Gefitinib (ZD1839 Iressa) is a low–molecular-weight quinazoline derivative that inhibits the activation of EGFR tyrosine kinase via competitive binding of the ATP-binding domain of the receptor. In a Gynecologic Oncology Group clinical trial, 27 patients with recurrent or persistent epithelial ovarian cancer were treated with 500-mg gefitinib daily (43). Four patients (14.8%) survived progression free for more than 6 months, with one objective response (3.6%). Commonly observed toxicity included skin rash and diarrhea. Interestingly, EGFR expression was associated with longer progression-free survival and possibly longer survival. The patient with the only objective antitumor response had a tumor with a mutation in the catalytic domain of the tumor’s EGFR (2235dEL15). This patient received 29 cycles of gefitinib and had a progression-free survival of approximately 27 months.
In a separate trial, 24 patients with recurrent epithelial ovarian cancer were treated with gefitinib 500 mg daily (44). All tumor samples had detectable levels of EGFR and PGFR. Of 16 patients who completed more than two cycles of therapy, no complete or partial responses were observed. However, analysis of clinical samples showed that gefitinib inhibited phosphorylation of EGFR, thereby providing a conceptual proof of targeted therapy.
Treatment of patients with recurrent ovarian cancer using the combination of gefitinib, carboplatin, and paxitaxel has resulted in an overall response rate of 63% (45). Interestingly, antitumor responses were observed in 35% of patients with platinum-resistant disease compared to 73% of patients with platinum-sensitive disease. Based on these preliminary data, none of the 18 patients treated showed EGFR receptor mutations.
Gefitinib has also been used in combination with tamoxifen. In 56 patients with primary ovarian or fallopian tube cancer, treatment with tamoxifen (40 mg/day) and gefitinib (500 mg/day) did not result in objective antitumor responses, but 16 patients had stable disease (46). In squamous and adenocarcinoma of the cervix, gefitinib (500 mg/day) treatment resulted in disease stabilization in 6 of 28 patients (20%) but no clinical responses (47). The median duration of stable disease was 111.5 days, with a median overall survival of 107 days.
Lapatinib is a small-molecule inhibitor of both the HER2 and EGFR tyrosine kinase receptor. The rationale for using lapatinib in endometrial carcinoma is supported mainly by studies in human cancer cell lines. Its efficacy in endometrial cancer is being investigated currently in clinical trials (33).
HER2/neu
The HER2/neu receptor is activated by homo- or heterodimerization, resulting in tyrosine phosphorylation and subsequent activation of various downstream signals that among other functions control cellular proliferation, migration, and invasion. Trastuzumab is a recombinant, humanized IgG1 monoclonal antibody that is specific for the extracellular domain of HER2/neu. Binding of the antibody to HER2/neu prevents activation of the receptor with a subsequent increase of apoptosis in vitro and in vivo, impaired DNA damage repair, and inhibition of tumor neovascularization (35). Preclinical models have suggested that the therapeutic activity may also depend on innate immune effector cells that mediate antibody-dependent cellular cytotoxicity (ADCC). In addition, trastuzumab influences the adaptive immune response and augments antigen processing and presentation (48). In breast cancer, the addition of trastuzumab to adjuvant chemotherapy for patients with HER2/neu-positive tumors significantly decreases the hazard ratio for recurrence and subsequently improves survival (49).
The HER2/neu oncogene is overexpressed in several gynecologic malignancies, including 20% to 30% of ovarian cancers (50). The largest clinical trial evaluating HER2/neu as a target in ovarian or peritoneal carcinoma was conducted by the Gynecologic Oncology Group (51). Of 837 tumor samples screened for HER2/neu expression, 95 patients (11.4%) were found to have tumors with HER/neu overexpression. Forty-one patients with HER2/neu-positive tumors received trastuzumab weekly. Single agent treatment resulted in one complete (2.4%) and two partial (4.9%) responses, with a median duration of response of 8 weeks (range: 2 to 104 weeks). The authors concluded that single agent trastuzumab in recurrent ovarian cancer was of limited value because of the low frequency of HER2/neu overexpression and the low rate of clinical antitumor response.
HER2/neu overexpression is infrequent in cervical cancer. In one study, only one of 35 (2.9%) cervical carcinomas showed strong expression of HER2/neu (52). In uterine serous carcinoma, 12 of 68 tumors (18%) showed HER2/neu overexpression; this was associated with a worse overall prognosis (53). In a separate study, 5 of 19 specimens (26%) stained strongly for HER2/neu protein receptor (54).
Mitogen-activated Protein Kinase Pathways
The mitogen-activated protein (MAP) kinase cascades are activated by various cofactors, inflammatory cytokines, and stress (55). The signaling cascades include various molecules, including RAS, MEK1/2, ERK1/2, and p38 MAPK. Various molecules have been developed that target this pathway but are mostly still under investigation. Sorafenib is among the first of the agents with clinically proven efficacy. Sorafenib is a competitive inhibitor of raf that has been approved for treatment of renal cell carcinoma and hepatocellular carcinoma (56). Besides targeting raf, sorafenib also inhibits VEGFR2 and VEGFR3, FT3, c-kit, and PDGFR-β.
Targeting the MAPKinase pathway has been a particular focus of clinical studies in low-grade serous ovarian carcinomas. The latter are characterized by younger age at presentation, indolent growth pattern, and poor response to systemic therapy (57). Up to one-third of low-grade serous ovarian carcinomas have been found to have a mutation in either BRAF or KRAS, and hence activated MAPKinase pathway signaling (58). A phase II trial of the MEK1/2 inhibitor selumetinib (AZD6244) in 52 patients with recurrent low-grade serous ovarian carcinoma showed an overall response rate of 15.4%, stable disease in 65% of patients, and a PFS of 11 months (59). The 6% BRAF, 41% KRAS, and 15% NRAS mutations did not correlate with response to therapy.
PARP Inhibitors
The enzyme poly (adenosine diphosphate-ribose) polymerase (PARP) and the BRCA proteins are involved in DNA repair as induced by cytotoxic agents like platinum chemotherapy or radiation. The recent development of PARP inhibitors has allowed the therapeutic targeting of DNA repair pathways and exploited the concept of synthetic lethality. In essence, in a synthetic lethal pair, targeting of one gene product or protein (PARP) while the other gene is defective (BRCA mutation) selectively kills tumor cells while sparing normal cells (60). It has therefore been hypothesized that patients harboring mutations in BRCA1–2 would be highly susceptible to treatment with PARP inhibitors. Several clinical trials have documented proof of this mechanism, including a large phase I study of the PARP inhibitor olaparib in women with BRCA1 or BRCA2 germline mutations (61,62). In a randomized phase II trial, patients with platinum-sensitive recurrent ovarian cancer who achieved a response to platinum-based chemotherapy were treated with maintenance olaparib (63). Among the 265 women, 22.8% had BRCA mutations, while 13.2% were wild type and 64% unknown. Treatment with olaparib at a dose of 400 mg twice daily resulted in a significantly longer median PFS compared to a placebo (median: 8.4 months vs. 4.8 months; HR = 0.35; 95% CI 0.25 to 0.49). Similarly, a randomized phase II trial of paclitaxel–carboplatin with or without olaparib followed by olaparib or placebo maintenance showed a significant prolongation of the median PFS in the experimental arm (median: 12.2 months vs. 9.6 months; HR = 0.51; 95% CI 0.34 to 0.77) (64). PARP inhibitors are currently being extensively studied in various clinical trials and include olaparib, niraparib, BMN-673, rucaparib, and veliparib. The efficacy of PARP inhibitors might not be restricted to patients with mutated BRCA, but may also be seen in tumors with functional defects in other DNA repair pathway proteins.
The PI3-kinase/Akt/mTOR Pathway
The phosphoinositide3-kinase (PI3-kinase)/Akt/mammalian target of rapamycin (mTOR) pathway is a major oncogenic signaling pathway in various cancers (65). Activation of this pathway can be demonstrated in more than 80% of endometrial cancers, 50% to 70% of epithelial ovarian cancers, and approximately 50% of cervical cancers (66–68). Activation of PI3-kinase by various growth factors such as platelet-derived growth factor (PDGF) or insulin growth factor results in phosphorylation and therefore activation of the central oncogenic protein Akt. Activated Akt is released from the membrane and elicits downstream effects, mainly by phosphorylating signal transduction proteins such as BAD, FKHR, Caspase 9, and the mTOR. Activation of these downstream signals leads to an increase in cellular proliferation, invasiveness, drug resistance, and neoangiogenesis. The PTEN gene (phosphatase and tensin homolog deleted on chromosome 10) is a tumor suppressor gene that is located on chromosome 10q23 and encodes a dual-specificity phosphatase for both lipid and protein substrates (69). PTEN decreases the activation of Akt in the PTEN/PI3-kinase/Akt pathway.
Several inhibitors of PI3-kinase/Akt/mTOR signaling are currently in clinical trials (70,71). Rapamycin or rapamycin analogs, for example, block the activity of mTOR, a protein complex responsible for increasing protein synthesis and cellular proliferation (72). Several mTOR inhibitors, including RAD001 and CCI779, and specific PI3-kinase inhibitors are currently under development in preclinical models and clinical trials. PI3-kinase/Akt/mTOR inhibitors have been used in endometrial cancer with limited benefit (73). However, the results from clinical trials using mTOR inhibitors in renal cell carcinomas and glioblastomas are encouraging (74).
Inhibitors of the PI3K pathway signaling might have greater antitumor effects in combination with other targeting strategies, including PARP inhibition or targeting VEGF, based on the following rationale: PI3K inhibition is associated with the loss of homologous recombinant repair capability resulting in sensitization to PARP inhibitors. PARP inhibitors can increase VEGFR2 phosphorylation and the subsequent activation of endothelial cell survival pathways that can be blocked with antiangiogenic therapies.
Immunotherapy
Failure of functional immunity contributes to the genesis of virus-associated cancers, such as those caused by human papillomavirus (HPV) or Epstein–Barr virus. Although many effective induced antitumor immune responses have been described, the relative role of natural antitumor immune responses in the detection and destruction of cancer cells, at least as was envisioned originally when the concept of immune surveillance was first defined (76), is still unclear. Some researchers have suggested that immune responses are mainly involved in protection from virus-associated cancers, but not from other forms of cancer (77).
Cancer is a common disease, and overt immune deficiency certainly is not necessary for its development. However, recent studies have shown that many cancers, including those that are not known to have a viral etiology, are seen with increased frequency in patients who have dysfunctional immunity. In a meta-analysis of cancer incidence in populations known to be immune deficient (e.g., organ transplant recipients, patients with HIV infection), Grulich et al. (78) found an increased incidence of several common cancers, suggesting that impaired immunity may contribute to the development of cancer.
Components of the Immune System Involved in Antitumor Responses
Various types of human immune responses can target tumor cells. Immune responses can be categorized as humoral or cellular, a distinction based on the observation in experimental systems that some immune responses could be transferred by serum (humoral) and others by cells (cellular). In general, humoral responses refer to antibody responses; antibodies are antigen-reactive, soluble, bifunctional molecules composed of specific antigen-binding sites associated with a constant region that directs the biologic activities of the antibody molecule, such as binding to effector cells or complement activation (Fig. 2.2). Cellular immune responses generally refer to cytotoxic responses mediated directly by activated immune cells, rather than by the production of antibodies (Fig. 2.3).
Nearly all immune responses involve both humoral and cellular components and require the coordinated activities of populations of lymphocytes operating in concert with each other and with antigen-presenting cells. These activities result in various effector functions such as antibody production, cytokine secretion, and the stimulation and expansion of cytotoxic T cells. Cellular interactions involved in immune responses include direct cell–cell contact as well as cellular interactions mediated by the secretion of, and response to, cytokines. The latter are biologic messenger molecules that play important roles in the genesis, amplification, and effector functions of immune responses.
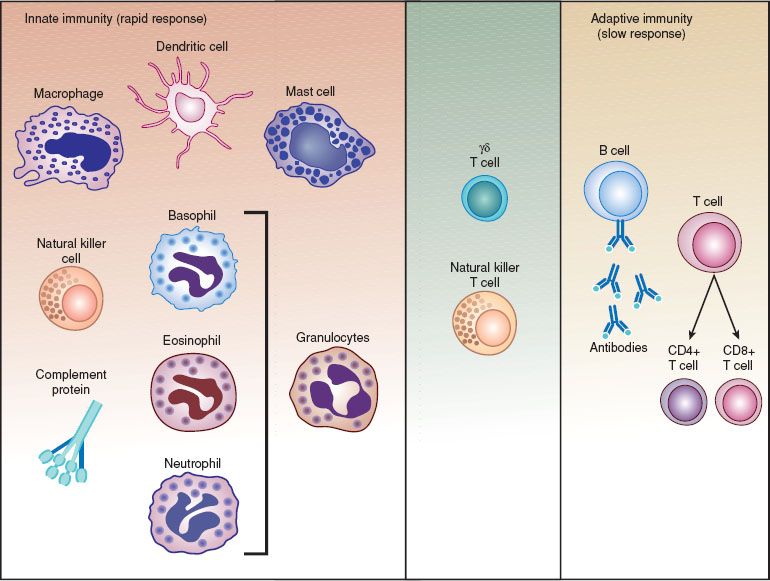
Figure 2.2 The components of the immune system.
T lymphocytes play a pivotal role by acting as helper cells in the generation of humoral and cellular immune responses and by acting as effector cells in cellular responses. Cytotoxic T cells are effector T cells that can directly interact with, and kill, target cells by the release of cytotoxic molecules and the induction of target cell apoptosis. T lymphocyte precursors mature into functional T lymphocytes in the thymus, where they learn to recognize antigen in the context of the major histocompatibility complex (MHC) molecules of the individual. Most T lymphocytes with the capability of responding to self-antigens are removed during thymic development. T lymphocytes are distinguished from other types of lymphocytes by their biologic activities and by the expression of distinctive cell surface molecules, including the T cell antigen receptor and the CD3 molecular complex. T lymphocytes recognize specific antigens by interactions that involve the T cell antigen receptor (Fig. 2.2) (79).
There are two major subsets of T lymphocytes: T helper/inducer cells, which express the CD4 cell surface marker; and T suppressor/cytotoxic cells, which express the CD8 marker. CD4 T lymphocytes can provide help to B lymphocytes, resulting in antibody production, and also can act as helper cells for other T lymphocytes. Much of the helper activity of T lymphocytes is mediated by the production of cytokines. CD4 T cells have been further subdivided into TH1 (cellular immunity/proinflammatory) and TH2 (antibody response–promoting) subsets, based on the patterns of cytokine production and the biologic properties of these cells. Recent studies have identified a subset of T cells that inhibit autoreactive cells, perhaps acting to prevent autoimmune responses (80). This subset of T cells has been called regulatory T (Treg) cells. Other recently described T cell subsets include TH17 cells, which are important in driving responses to bacteria and fungi (81,82).
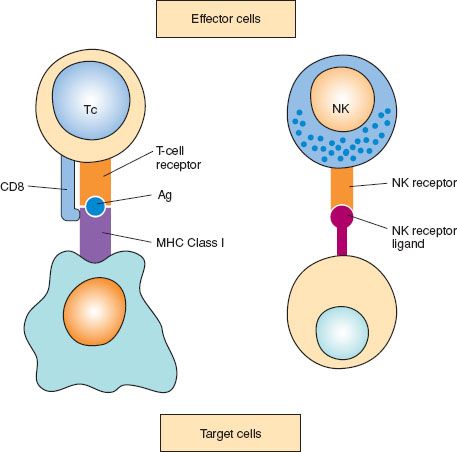
Figure 2.3 Cell-mediated cytotoxicity: two different types: 1. Cytotoxic T cells (Tc) bind to their target by recognizing specific antigens (Ag) in the context of major histocompatibility complex (MHC) determinants. 2. Natural killer (NK) cells recognize target cells in an unspecific manner via NK receptor ligands.
The CD8 T lymphocyte subset includes cells that are cytotoxic and can directly kill target cells. A major biologic role of such cytotoxic T lymphocytes is the lysis of virus-infected cells. Cytotoxic T lymphocytes can directly mediate the lysis of tumor cells. Effector T cells also can contribute to antitumor immune responses by producing cytokines, such as tumor necrosis factor (TNF), that induce tumor cell lysis and can enhance other antitumor cell effector responses.
Both CD4 and CD8 T cells respond to antigen only when it is presented in the context of major histocompatibility complex (MHC) molecules on antigen-presenting cells, target cells, or both. The T cell receptor on CD4 T cells is restricted to responding to antigen plus MHC class II molecules; the receptor on CD8 T cells is restricted to responding to antigen plus MHC class I molecules. In addition, both T cell subsets require a second simultaneous costimulatory signal for optimal stimulation, in the absence of which the T cells may be induced to enter a state of unresponsiveness or even apoptosis. Therefore, provision of effective costimulatory signals is necessary for the induction of effective antitumor responses by activated T cells.
In ovarian cancer, the presence of tumor-infiltrating lymphocytes (TILs) correlates both with progression-free and overall survival (83,84). In particular CD8-positive TILs are prognostic markers, since their presence correlates with survival across all stages of disease and all histologic types. In contrast, the presence of immunosuppressive Treg cells (classified as CD4+/CD25+/FoxP3+ T cells), in ovarian cancers has been associated with decreased survival (85,86).
B lymphocytes are the cells that produce and secrete antibodies, which are antigen-binding molecules (Fig. 2.2). B lymphocytes develop from pre-B cells and, after exposure to antigen and appropriate activation signals, differentiate to become plasma cells—cells that produce large quantities of antibodies. Mature B lymphocytes use cell-surface immunoglobulin molecules as antigen receptors.
In addition to producing antibodies, B lymphocytes play another important role: they can serve as efficient antigen-presenting cells for T lymphocytes. Although the production of antitumor antibodies does not appear to play a central role in host antitumor immune responses, monoclonal antibodies reactive with tumor-associated antigens have proven to be very useful in antitumor therapy, as well as in the detection of tumors or of tumor-associated molecules. Unfortunately, no truly unique tumor-specific antigens have been identified, and most tumor-related antigens are also expressed to some extent on nonmalignant tissues.
Because some monoclonal antibodies are of murine and not human origin, the host’s immune system can recognize and respond to murine monoclonal antibodies. This has led to the development of “humanized” monoclonal antibodies (genetically engineered monoclonal antibodies composed of human constant regions with specific antigen-reactive murine variable regions), with the aim of avoiding many of the problems associated with the administration of murine monoclonal antibodies.
Macrophages and dendritic cells (DCs) also play key roles in the generation of adaptive, lymphocyte-mediated immune responses by acting as antigen-presenting cells. Helper/inducer (CD4) T lymphocytes, bearing a T cell receptor of appropriate antigen and self-specificity, are activated by antigen-presenting cells that display processed antigen combined with self-MHC molecules (Fig. 2.2). Antigen-presenting cells also provide costimulatory signals that are important for the induction of T lymphocyte activation. In addition to serving as antigen-presenting cells, macrophages can ingest and kill microorganisms, and act as cytotoxic antitumor killer cells. These cells also produce various cytokines, including IL-1, IL-6, chemokines, IL-10, and TNF, which are involved in many immune responses. These monocyte-produced cytokines can have direct effects on tumor cell growth and development, both as growth-inducing and growth-inhibiting factors.
Natural killer (NK) cells are cells that have large granular lymphocytic morphology, do not express the CD3 T cell receptor complex, and do not respond to specific antigens. NK cells can lyse target cells, including tumor cells, unrestricted by the expression of antigen or self-MHC molecules on the target cell. Therefore, NK cells are effector cells in an innate (nonantigen-restricted) immune response, and may play a vital role in immune responses to tumor cells. The cells that can affect ADCC are NK-like cells.
Cytokines are soluble mediator molecules that induce, enhance, or affect immune responses. Cytokines are produced by various types of cells and play critical roles not only in immune responses, but also in biologic responses outside of the immune response, such as hematopoiesis or the acute phase response. T helper 1 (TH1) and TH2 cells, which control the nature of an immune response by secreting characteristic and mutually antagonistic sets of cytokines (9–11), are defined by the cytokines they produce. TH1 clones produce IL-2 and IFN-γ, whereas TH2 clones produce IL-4, IL-5, IL-6, and IL-10. TH1 cytokines promote cell-mediated and inflammatory responses, whereas TH2 cytokines enhance antibody production. Most immune responses involve both TH1 and TH2 components.
Research has identified CD4-positive T cells that participate in the maintenance of immunologic self-tolerance by actively suppressing the activation and expansion of self-reactive lymphocytes. These cells are called Treg cells. Treg cells are characterized by the expression of CD25 (the IL-2 receptor chain) and the transcription factor FoxP3 (87,88). Treg cell activity is thought to be important in preventing the development of autoimmune diseases. Removal of Treg also may enhance immune responses against infectious agents or cancer. Although much remains to be learned about the role of Treg activity in antitumor immunity, it is clear that such cells may play a role in modulating host responses to cancer.
Therapeutic Strategies
There has been great interest in developing effective biologic and immunologic therapies for gynecologic malignancies. For example, patients with small-volume or microscopic residual peritoneal ovarian cancer are attractive candidates for peritoneal immune or biologic therapy (89,90). Also, many patients with advanced disease are immunocompromised, suggesting a role for immune-enhancing therapeutic approaches. Advances in molecular biology, biotechnology, immunology, and cytokine biology have resulted in the availability of many new, promising immunotherapeutic approaches for gynecologic cancers.
The state of the art in immunotherapy for ovarian cancer and other gynecologic malignancies has been discussed in detail in several recent reviews (91–94), and the reader is referred to these publications for more detailed information. Examples of the current use of immunotherapy in clinical trials are provided below.
Cancer Vaccines
Cancers may develop or progress because immune cells are not given a strong enough signal to become activated and destroy the tumor cells. In some cases, cancers are able to downregulate immune responses. For example, cytokines or other molecules produced by tumor cells, such as IL-10 (132), are able to inhibit antitumor immune responses. It may be possible to counter this lack of antitumor immune responsiveness by enhancing antigen presenting cell (APC) activity, providing tumor-associated antigens in a manner that can better induce the generation of antitumor effector T cells (tumor vaccine therapy), or both.
Cervical Cancer
The greatest success story involving the enhancement of immunity to combat gynecologic cancer is the development of vaccines against human papilloma virus (HPV), which are highly effective for the prevention of cervical dysplasia and cancer (94). In addition, dysplastic and cancerous cervical epithelial cells infected with HPV, an oncogenic virus, also present an attractive target for immune enhancement-based therapeutic strategies, including the development of therapeutic vaccines for HPV.
HPV—specifically, HPV subtypes 16, 18, 31, and 45—has been implicated as the major etiologic agent in cervical cancer. HPV-infected dysplastic and cancerous cervical epithelial cells consistently retain and express two of the viral genes, E6 and E7, that respectively interact with, and disrupt, the function of the p53 and retinoblastoma tumor suppressor gene products. Factors other than infection with HPV, such as cellular immune function, play an important role in determining whether the infection of cervical epithelial cells regresses or progresses to cancer. This has led to the development of prophylactic and therapeutic vaccines to HPV as well as treatment approaches based on the enhancement of host immune function.
HPV vaccines have been shown to have an exceptional level of efficacy (75), clearly reducing the incidence of both HPV-16 and HPV-18 infections, and HPV-16 and HPV-18–related cervical intraepithelial neoplasia. The HPV vaccines Gardasil and Cervarix use HPV-like particles as immunogens to generate neutralizing antibodies for HPV. HPV-based therapeutic cancer vaccines may ultimately be effective for the control of cervical cancer (94).
HPV E6 and E7 are attractive antigens for use in therapeutic vaccines because these HPV-encoded proteins are involved in cellular transformation, and therefore are consistently expressed in HPV-positive tumor cells. Candidate therapeutic HPV vaccines include DNA vaccines, with recent research aimed at enhancing the potency and delivery of such vaccines (94,134–136). Clinical trials with such DNA-based vaccines are currently being planned (94).
Ovarian Cancer
Therapeutic vaccines for ovarian cancer have been developed and studied in various clinical trials. A thorough overview of the rationale and design of potential vaccines can be found in recent review papers (91,92). At this time, tumor vaccine approaches include (i) vaccination with defined tumor-associated antigens, or DNA vaccines that encode for tumor-associated antigens; and (ii) vaccination with whole tumor cell preparations, with and without the coadministration of antigen-presenting cells such as dendritic cells (DCs).
Peptide vaccines are designed to stimulate antitumor immune responses against various target antigens that are expressed on ovarian cancer, such as NY-ESO-1, p53, WT-1, HER-2, and VEGF (133). These vaccines are generally well tolerated and can generate measurable, sustained immune responses (95). Peptide vaccines are frequently administered with adjuvants, such as Montanide or granulocyte–macrophage colony-stimulating factor (GM-CSF) to enhance immune responses in vivo.
One of the candidate antigen for vaccine development is NY-ESO-1 which is a highly immunogenic cancer-testis antigen. It is expressed in up to 40% of ovarian cancers (96). Two recent clinical trials have shown the ability of an NY-ESO-1 peptide vaccine with Montanide ISA51 as adjuvant to induce both antigen-specific CD4+ and CD8+ T cell responses in patients with minimal residual ovarian cancer (97). The second trial used HLA-A*0201-restricted NY-ESO-1b peptide vaccination with Montanide in patients with ovarian cancer in complete clinical remission after first-line treatment (98). Three of four patients with NY-ESO-1-positive tumors and four of five patients with NY-ESO-1-negative tumors showed T cell immunity. Three patients with NY-ESO-1-negative tumors remained in complete clinical remission. Another trial used intradermally injected recombinant vaccinia and fowlpox viruses expressing NY-ESO-1 in 19 women with NY-ESO-1 expressing ovarian cancers in complete remission after primary therapy (50). About 50% of patients showed NY-ESO-1-specific immune responses, resulting in a mean disease-free interval of 19.9 months (99).
Other vaccination strategies have used p53 peptides as the immunogenic antigen, since the vast majority of serous cancers in particular overexpress p53 (100–102). In general, p53 vaccination trials have resulted in the successful generation of p53-specific immune responses, and the vaccines have been well tolerated. Clinical responses have been demonstrated in up to 20% of patients, but these responses have not necessarily correlated with vaccine-mediated antip53 immunity.
Various multipeptide vaccines have been studied in ovarian cancer patients. One of these vaccines combines peptides from various antigens, including the adenomatous polyposis coli, ubiquitin-conjugating enzyme E2, BAP31, replication protein A, Abl-binding protein 3c, cyclin I, topoisomerase IIα, integrin β eight subunit precursor, cell division control protein 2 (CDC2), TACE/ADAM17, g-catenin, and EDDR1, administered along with Montanide ISA-51 and GM-CSF. This vaccine generated peptide-specific T cell responses in patients with breast and ovarian cancer without evidence of disease, but 50% of patients with ovarian cancer recurred during the observation period (103).
Recombinant vaccinia and fowlpox viruses have been used to express CEA and MUC1 as well as the T cell costimulatory molecules B7.1, ICAM-1, and LFA-3. The vaccine showed efficacy in one of the three ovarian cancer patients treated in a pilot study that enrolled patients with MUC1-positive cancers (104).
In addition to using specific tumor-associated antigen, whole tumor antigen vaccines can potentially provide a wider range of tumor antigens. These vaccines can be created using autologous tumor lysates or tumor-derived RNA, and are in general administered with adjuvants like GM-CSF, Montanide ISA-51, or toll-like receptor agonists. Patients may have a better clinical response to whole tumor antigen vaccination (105,106).
Dendritic cells are highly effective antigen-presenting cells, and play a central role in the induction of both CD4 and CD8 T cell responses. DCs can be pulsed with tumor antigen peptides, or bioengineered to express tumor antigens, allowing them to be used in experimental therapies that aim to enhance antitumor immunity. Exposure of T cells to DCs pulsed with ovarian cancer–derived antigenic preparations has resulted in the generation of cytolytic effector T cells that are capable of killing autologous tumor cells in vitro (137–139).
In a phase I clinical trial, Hernando et al. (140) showed that patients with advanced gynecologic malignancies could be effectively vaccinated with DCs pulsed with a nontumor test antigen, keyhole limpet hemocyanin (KLH), and autologous tumor antigens. Lymphoproliferative responses to KLH and to tumor lysate stimulation were noted. The treatment was safe, well tolerated, immunologically active, and generally devoid of significant adverse effects.
Given the exceptional ability of DCs to serve as potent antigen-presenting cells in the induction of T cell responses, an attractive approach would be to combine tumor vaccines with ex vivo–generated DC preparations. DCs can be generated ex vivo from peripheral blood mononuclear cells, using various strategies (91,141–144). Such ex vivo–generated DCs can be pulsed with tumor antigens or vaccines, or with DNA- or RNA-encoding tumor antigens before administration, and have resulted in the induction of antitumor responses in preclinical studies (91,145–147).
Several major challenges need to be overcome for the successful development of effective DC-based therapies for ovarian cancer: (i) The identification of tumor-associated or tumor-specific antigens, (ii) the development of means to induce optimal DC maturation after antigen uptake, (iii) the development of schemes for generation of DCs that maintain optimal antigen-presenting cell activity and do not produce immunosuppressive factors, and (iv) the development of ex vivo expansion techniques that provide sufficient numbers of DCs for effective immunotherapy (91,148–150). As more is learned about the immunogenicity of current tumor-associated antigens, novel cancer-associated antigens are identified, and the techniques of DC activation and antigen expression are better developed, DC-based immunotherapy may provide a therapeutic alternative for the treatment of these cancers.
Monoclonal Antibodies and Antibody-based Immunotherapy
Monoclonal antibodies have played an important role in both the development of immunotherapeutic agents and tumor markers. Monoclonal antibodies also have been used for radioimmunodetection (107,108) and are being used for treatment. Monoclonal antibodies can potentially induce antitumor responses in various ways: (i) By complement activation and subsequent tumor cell lysis; (ii) by directly inducing antiproliferative effects, perhaps by interaction with tumor cell surface signaling molecules; (iii) by enhancing the activity of phagocytic cells, which can interact with immune complexes containing monoclonal antibodies; and (iv) by mediating ADCC via interactions of the Fc portion of monoclonal antibodies with Fc receptors on cells that mediate ADCC (109). In addition, monoclonal antibodies can be labeled with either radioactive particles or antitumor drugs and used to focus these agents onto tumor cells (110).
Some monoclonal antibody-based drugs are currently approved for the treatment of cancer. FDA-approved monoclonal antibody-based anticancer drugs include bevacizumab (Avastin) for the treatment of colon and lung cancer, cetuximab (Erbitux) for the treatment of colon and head and neck cancer, gemtuzumab (Mylotarg) for the treatment of acute myelogenous leukemia, rituximab (Rituxan) for the treatment of nonHodgkin lymphoma, and trastuzumab (Herceptin) for the treatment of breast cancer.
Various antigens have been found to be expressed by ovarian cancers and are being explored as therapeutic targets (111). In general, these antigens can be classified as specific tumor-associated antigens or universal tumor antigens. Examples of ovarian tumor-associated antigens include CA125, the folate receptor, MUC1, mesothelin, and NY-ESO-1. Survivin and hTERT are universal tumor antigens, etc., since they are expressed on a variety of tumors, and not on most normal human cells.
Several clinical trials have utilized monoclonal antibodies directed against ovarian cancer antigens, including CA125, folate receptor, MUC1 antigen, and tumor-associated glycoprotein 72 (93). Evidence that CA125 can act as a tumor antigen and stimulate humoral and cellular immune responses has been derived from various in vitro studies and clinical trials. Oregovomab (B43.13) is a murine monoclonal antibody to CA125 that has been used for the treatment of ovarian cancer. The antibody binds to circulating CA125, resulting in the formation of immune complexes (antibody–antigen complexes). These immune complexes are recognized as foreign, mainly because of the murine component. They are taken up by antigen-presenting cells, allowing the processing of the autologous CA125 antigen, ultimately leading to induction of CA125-specific antibodies, helper T cells, and cytolytic T cells.
In 2004, Berek et al. reported on the use of oregovomab for maintenance therapy in patients with ovarian cancer after first-line treatment. A subgroup of patients with favorable prognostic factors had a significantly longer time to relapse compared to patients in the placebo group (112). This subgroup was studied in a subsequent trial and oregovomab maintenance therapy failed to show a survival benefit (113,114). Oregovomab is currently being studied in combination with carboplatin and paclitaxel, and might provide immune adjuvant properties in this setting, as suggested by recent clinical observations (115).
Another antibody network–based strategy has employed anti-idiotypic vaccines in patients with relapsed ovarian cancer. ACA125 is a murine anti-idiotypic antibody that mimics an antigenic epitope on CA125 (92,93,116,117). Therefore, antibodies generated to ACA125 have the potential to react with antigenic epitopes on CA125, with ACA125 serving as an anti-idiotypic vaccine that would enhance immune responses to CA125 (92). Treatment with ACA125 has resulted in both humoral and cellular responses, and those patients who had detectable anti-ACA125 responses showed a longer mean survival time (118,119).
Abagovomab is an anti-idiotypic antibody that mimics the CA125 antigen. The initial results of abagovomab treatment in patients with ovarian cancer have been reported by Sabbatini et al. (120) and have shown that all patients developed an anti-idiotypic antibody response (Ab3). In addition, the generation of T cell immunity to CA125 was demonstrated in five patients. While patients had measurable serum CA125 levels in both trials, neither trial analyzed CA125 expression in tumor tissue. A large international, multicenter trial is investigating the effect of abagovomab as consolidation treatment in patients with ovarian cancer. Preliminary data from the trial does not show a clinical benefit of abagovomab as consolidation therapy.
Adoptive lmmunotherapy
Adoptive immunotherapy involves the ex vivo expansion of antitumor immune cells followed by the administration of such effector cells. It has provided another immune system–based approach for antitumor therapy (91,92,121–124). Adoptive immunotherapy, involving the infusion of large numbers of autologous ex vivo–activated immune effector cells, has been shown to produce tumor regression in various animal and human tumors (122), and it has produced the best results to date in tumor immunotherapy (91). This approach can provide large numbers of tumor-specific T cells with the capacity to specifically kill tumor cells, and can potentially lead to the complete elimination of residual tumor cells (91).
Early approaches used peripheral blood mononuclear cells exposed to IL-2 ex vivo to lead to the generation of lymphokine-activated killer (LAK) cells that are cytotoxic for a variety of tumor cells (125,126). Although experimental treatment of human subjects with LAK cells and IL-2 yielded some responses, considerable toxicity was seen (89,121–124,126–130), and adoptive immunotherapy with LAK cells does not appear to be a practical option for the treatment of ovarian cancer.
The use of immunotherapy based on ex vivo–stimulated tumor-infiltrating lymphocytes or tumor-associated lymphocytes from ascites, with or without added IL-2, also has been examined in ovarian cancer (91,124,129,130). It is clear that optimization of such adoptive immunotherapies is needed in terms of the cell source, the forms of stimulation, the methods for ex vivo expansion, and the cytokines that are given during such treatment (91). Potentially important refinements of these approaches include the use of DCs as antigen-presenting cells (APCs) to stimulate T cells, the provision of effective costimulatory signals to the responding T cells by APC, host conditioning with immunosuppressive chemotherapy before the adoptive transfer of cells (91,131), and the genetic modification of T cells to express tumor-specific antigens like NY-ESO-1.
Cytokine Therapy: Modulation of Host Immunity
Most early experimental biologic therapies for metastatic ovarian cancer involved biologic response modifiers such as Corynebacterium parvum (a heat-killed, gram-negative anaerobic bacillus), bacillus Calmette–Guérin (BCG), or modifications of these agents (152–155). Intraperitoneal (IP) treatment with C. parvum induced a profound local reaction, including peritoneal fibrosis, and its toxicity precluded more widespread testing.
Malignancies that tend predominantly to grow in the peritoneal cavity, such as ovarian cancer, have been treated in many experimental trials with IP administered drugs, most frequently with cytotoxic chemotherapeutic agents (156). IP biologic response modifier therapy, immunotherapy with cytokines, and gene therapy have been proposed. These approaches have the additional advantage of potentially inducing the activation of regional immune effector mechanisms in the peritoneal cavity (90). This might be particularly true for cytokine-based treatment strategies or for adoptive immunotherapies, because activated immune effector cells may require direct contact with the malignant target cells for most effective antitumor activity.
Various cytokines have been tested in clinical trials, to date producing mixed results (91). This includes trials of IFN-α and IFN-f, TNF-α, and IL-2. IP IL-2 therapy was studied in women with platinum-resistant or refractory ovarian cancer in a phase I/II trial (157,158). Among 35 assessable patients, there were 6 complete responses and 3 partial responses, for an overall response rate of 25.7% (9 of 35) with a median survival of 2.1 years.
Treatment with IP IFN-α combined with cisplatin or carboplatin has resulted in pathologic complete and long-term remission in 14 of 27 patients in two trials for recurrent or refractory ovarian cancer (159,160). Treatment of patients with refractory ovarian cancer with intravenous recombinant human IL-12, a TH1-inducing cytokine, has resulted in disease stabilization in about half of the treated patients (161).
IP treatment with IL-12 in patients with carcinomatosis from mesotheliomas, Müllerian, or gastrointestinal carcinomas has shown disease stabilization in 2 of 12 treated patients (162). In a phase II trial, treatment with subcutaneously administered IL-2 and oral retinoic acid was reported to improve survival in 44 patients who had ovarian cancer responding to chemotherapy (163).
GM-CSF stimulates the proliferation and differentiation of granulocytes and monocytes, and has demonstrated antitumor efficacy in patients with recurrent Müllerian malignancies. In one clinical trial that included 72 patients, GM-CSF treatment resulted in one complete response and 20 patients had stable disease (164). GM-CSF in combination with interferon gamma 1b (IFN-γ 1b) and carboplatin in women with recurrent, platinum-sensitive ovarian cancer (165). Of 54 patients evaluable for response, 9 (17%) had a complete response, 21 (39%) had a partial response, and 24 (44%) had progressive disease. The overall response rate was 56%. With a median follow-up of 6.4 months, the PFS was 6 months.
The identification of T cell subpopulations that have potent immunoregulatory properties, such as Treg and TH17 cells, has provided new opportunities for the design of host immune system–modulating therapies with the aim of enhancing immune responses to cancer. Treg cells are immunoinhibitory, and can inhibit the induction of cytotoxic T cells, and may thereby inhibit host antitumor immune responses. Increased levels of Treg cells in ascites, blood, and tumor have been seen in patients with advanced ovarian cancer (166). These Treg cells can inhibit antitumor immunity and promote tumor cell growth in ovarian cancer (85). Therefore, blocking the action of these Treg cells is an important target in the development of new immunotherapeutic approaches.
Studies aimed at blocking Treg activity in patients with cancer have been initiated using monoclonal antibodies targeting CD25, a cell-surface molecule commonly expressed on these cells. To date, these studies have not resulted in enhanced anticancer immune responses (167), and more refined approaches to targeting Treg need to be developed.
TH17 cells are another recently identified Treg cell subpopulation, characterized by the secretion of IL-17. They have the ability to modulate Treg activity (81,82,168). The role of TH17 cells in cancer has not been clearly defined. An animal study reported that provision of IL-2 in the tumor microenvironment was associated with decreased TH17 activity and increased Treg activity (168). Although much more work needs to be done to define the role of TH17 cells in downregulating Treg activity, and perhaps in enhancing antitumor responses, future experimental treatment strategies aimed at enhancing TH17 activity may be of value in enhancing antitumor immune responses.
References
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70.
2. Collinson FJ, Hall GD, Perren TJ, et al. Development of antiangiogenic agents for ovarian cancer. Expert Rev Anticancer Ther. 2008;8:21–32.
3. Ashouri S, Garcia AA. Current status of signal transduction modulators in the treatment of gynecologic malignancies. Curr Treat Options Oncol. 2007;8:383–392.
4. Markman M. The promise and perils of “targeted therapy” of advanced ovarian cancer. Oncology. 2008;74:1–6.
5. Murdoch D, Sager J. Will targeted therapy hold its promise? An evidence-based review. Curr Opin Oncol. 2008;20:104–111.
6. Goede V, Schmidt T, Kimmina S, et al. Analysis of blood vessel maturation processes during cyclic ovarian angiogenesis. Lab Invest. 1998;78:1385–1394.
7. Martin L, Schilder R. Novel approaches in advancing the treatment of epithelial ovarian cancer: The role of angiogenesis inhibition. J Clin Oncol. 2007;25:2894–2901.
8. Grothey A, Ellis LM. Targeting angiogenesis driven by vascular endothelial growth factors using antibody-based therapies. Cancer J. 2008;14:170–177.
9. Alvarez AA, Krigman HR, Whitaker RS, et al. The prognostic significance of angiogenesis in epithelial ovarian carcinoma. Clin Cancer Res. 1999;5:587–591.
10. Cohen MH, Gootenberg J, Keegan P, et al. FDA drug approval summary: Bevacizumab plus FOLFOX4 as second-line treatment of colorectal cancer. Oncologist. 2007;12:356–361.
11. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342.
12. http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab
13. Burger RA, Sill MW, Monk BJ, et al. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: A Gynecologic Oncology Group Study. J Clin Oncol. 2007;25:5165–5171.
14. Cannistra SA, Matulonis UA, Penson RT, et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol. 2007;25:5180–5186.
15. Burger RA, Brady MF, Bookman MA, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365:2473–2483.
16. Perren TJ, Swart AM, Pfisterer J, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365:2484–2496.
17. Aghajanian C, Blank SV, Goff BA, et al. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30:2039–2045.
18. Pujade-Lauraine E, Hilpert F, Weber B, et al. AURELIA: A randomized phase III trial evaluating bevacizumab (BEV) plus chemotherapy (CT) for platinum (PT)-resistant recurrent ovarian cancer (OC). J Clin Oncol. 2012;30(18 Suppl):LBA5002.
19. Karlan BY, Oza AM, Richardson GE, et al. Randomized, double-blind, placebo-controlled phase II study of AMG 386 combined with weekly paclitaxel in patients with recurrent ovarian cancer. J Clin Oncol. 2012;30:362–371.
20. Garcia AA, Hirte H, Fleming G, et al. Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: A trial of the California, Chicago, and Princess Margaret Hospital phase II consortia. J Clin Oncol. 2008;26:76–82.
21. Cohn DE, Valmadre S, Resnick KE, et al. Bevacizumab and weekly taxane chemotherapy demonstrates activity in refractory ovarian cancer. Gynecol Oncol. 2006;102:134–139.
22. Wright JD, Secord AA, Numnum TM, et al. A multi-institutional evaluation of factors predictive of toxicity and efficacy of bevacizumab for recurrent ovarian cancer. Int J Gynecol Cancer. 2008;18:400–406.
23. Badgwell BD, Camp ER, Feig B, et al. Management of bevacizumab-associated bowel perforation: A case series and review of the literature. Ann Oncol. 2008;19:577–582.
24. Auranen A, Grénman S. Radiation therapy and biological compounds for consolidation therapy in advanced ovarian cancer. Int J Gynecol Cancer. 2008;18(Suppl 1):44–46.
25. Lu C, Thaker PH, Lin YG, et al. Impact of vessel maturation on antiangiogenic therapy in ovarian cancer. Am J Obstet Gynecol. 2008; 198:477.e1–e9; discussion 477.e9–10.
26. Kumar A, Petri ET, Halmos B, et al. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J Clin Oncol. 2008;26:1742–1751.
27. Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65:1566–1584.
28. Psyrri A, Kassar M, Yu Z, et al. Effect of epidermal growth factor receptor expression level on survival in patients with epithelial ovarian cancer. Clin Cancer Res. 2005;11:8637–8643.
29. de Graeff P, Crijns AP, Ten Hoor KA, et al. The ErbB signalling pathway: Protein expression and prognostic value in epithelial ovarian cancer. Br J Cancer. 2008;99:341–349.
30. Khalifa MA, Abdoh AA, Mannel RS, et al. Prognostic utility of epidermal growth factor receptor overexpression in endometrial adenocarcinoma. Cancer. 1994;73:370–376.
31. Scambia G, Benedetti Panici P, Ferrandina G, et al. Significance of epidermal growth factor receptor expression in primary human endometrial cancer. Int J Cancer. 1994;56:26–30.
32. Wang D, Konishi I, Koshiyama M, et al. Expression of c-erbB-2 protein and epidermal growth receptor in endometrial carcinomas. Correlation with clinicopathologic and sex steroid receptor status. Cancer. 1993;72:2628–2637.
33. Konecny GE, Venkatesan N, Yang G, et al. Activity of lapatinib, a novel HER2 and EGFR dual kinase inhibitor in human endometrial cancer cells. Br J Cancer. 2008;98:1076–1084.
34. Vaidya AP, Parnes AD, Seiden MV. Rationale and clinical experience with epidermal growth factor receptor inhibitors in gynecologic malignancies. Curr Treat Options Oncol. 2005;6:103–114.
35. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51.
36. Attard G, Kitzen J, Blagden SP, et al. A phase Ib study of pertuzumab, a recombinant humanised antibody to HER2, and docetaxel in patients with advanced solid tumours. Br J Cancer. 2007;97:1338–1343.
37. Galizia G, Lieto E, De Vita F, et al. Cetuximab, a chimeric human mouse anti-epidermal growth factor receptor monoclonal antibody, in the treatment of human colorectal cancer. Oncogene. 2007;26:3654–3660.
38. Wang S, Guo P, Wang X, et al. Preclinical pharmacokinetic/pharmacodynamic models of gefitinib and the design of equivalent dosing regimens in EGFR wild-type and mutant tumor models. Mol Cancer Ther. 2008;7:407–417.
39. Gordon AN, Finkler N, Edwards RP, et al. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: Results from a phase II multicenter study. Int J Gynecol Cancer. 2005;15:785–792.
40. Vasey PA, Gore M, Wilson R, et al. A phase Ib trial of docetaxel, carboplatin, and erlotinib in ovarian, fallopian tube, and primary peritoneal cancers. Br J Cancer. 2008;98:1774–1780.
41. Secord AA, Blessing JA, Armstrong DK, et al. Phase II trial of cetuximab and carboplatin in relapsed platinum-sensitive ovarian cancer and evaluation of epidermal growth factor receptor expression: A Gynecologic Oncology Group study. Gynecol Oncol. 2008;108:493–499.
42. Konner J, Schilder RJ, DeRosa FA, et al. A phase II study of cetuximab/paclitaxel/carboplatin for the initial treatment of advanced-stage ovarian, primary peritoneal, or fallopian tube cancer. Gynecol Oncol. 2008;110:140–145.
43. Schilder RJ, Sill MW, Chen X, et al. Phase II study of gefitinib in patients with relapsed or persistent ovarian or primary peritoneal carcinoma and evaluation of epidermal growth factor receptor mutations and immunohistochemical expression: A Gynecologic Oncology Group Study. Clin Cancer Res. 2005;11:5539–5548.
44. Posadas EM, Liel MS, Kwitkowski V, et al. A phase II and pharmacodynamic study of gefitinib in patients with refractory or recurrent epithelial ovarian cancer. Cancer. 2007;109:1323–1330.
45. Lacroix L, Pautier P, Duvillard P, et al. Response of ovarian carcinomas to gefitinib-carboplatin-paclitaxel combination is not associated with EGFR kinase domain somatic mutations. Int J Cancer. 2006;118:1068–1069.
46. Wagner U, du Bois A, Pfisterer J, et al. Gefitinib in combination with tamoxifen in patients with ovarian cancer refractory or resistant to platinum-taxane based therapy—a phase II trial of the AGO Ovarian Cancer Study Group (AGO-OVAR 2.6). Gynecol Oncol. 2007;105:132–137.
47. Goncalves A, Fabbro M, Lhomme C, et al. A phase II trial to evaluate gefitinib as second- or third-line treatment in patients with recurring locoregionally advanced or metastatic cervical cancer. Gynecol Oncol. 2008;108:42–46.
48. Reich O, Liegl B, Tamussino K, et al. p185HER2 overexpression and HER2 oncogene amplification in recurrent vulvar Paget’s disease. Mod Pathol. 2005;18:354–357.
49. De Laurentiis M, Cancello G, Zinno L, et al. Targeting HER2 as a therapeutic strategy for breast cancer: A paradigmatic shift of drug development in oncology. Ann Oncol. 2005;16(suppl 4):iv7–13.
50. Tuefferd M, Couturier J, Penault-Llorca F, et al. HER2 status in ovarian carcinomas: A multicenter GINECO study of 320 patients. PLoS ONE. 2007;2:e1138.
51. Bookman MA, Darcy KM, Clarke-Pearson D, et al. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: A phase II trial of the Gynecologic Oncology Group. J Clin Oncol. 2003;21:283–290.
52. Chavez-Blanco A, Perez-Sanchez V, Gonzalez-Fierro A, et al. HER2 expression in cervical cancer as a potential therapeutic target. BMC Cancer. 2004;4:59.
53. Slomovitz BM, Broaddus RR, Burke TW, et al. Her-2/neu overexpression and amplification in uterine papillary serous carcinoma. J Clin Oncol. 2004;22:3126–3132.
54. Villella JA, Cohen S, Smith DH, et al. HER-2/neu overexpression in uterine papillary serous cancers and its possible therapeutic implications. Int J Gynecol Cancer. 2006;16:1897–1902.
55. McCubrey JA, Milella M, Tafuri A, et al. Targeting the Raf/MEK/ERK pathway with small-molecule inhibitors. Curr Opin Investig Drugs. 2008;9:614–630.
56. Adnane L, Trail PA, Taylor I, et al. Sorafenib (BAY 43–9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612.
57. Diaz-Padilla I, Malpica AL, Minig L, et al. Ovarian low-grade serous carcinoma: A comprehensive update. Gynecol Oncol. 2012;126:279–285.
58. Singer G, Oldt R III, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–486.
59. Farley J, Brady WE, Vathipadiekal V, et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: An open-label, single-arm, phase 2 study. Lancet Oncol. 2013;14:134–140.
60. Ashworth A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–3790.
61. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134.
62. Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet. 2010;376:245–251.
63. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392.
64. Oza AM, Cibula D, Oaknin A, et al. Olaparib plus paclitaxel and carboplatin (P/C) followed by olaparib maintenance treatment in patients (pts) with platinum-sensitive recurrent serous ovarian cancer (PSR SOC): A randomized, open-label phase II study [abstract]. J Clin Oncol. 2012;30(Suppl):a5001.
65. Jiang BH, Liu LZ. PI3 K/PTEN signaling in tumorigenesis and angiogenesis. Biochem Biophys Acta. 2008;1784:150–158.
66. Mutter GL, Lin MC, Fitzgerald JT, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst. 2000;92:924–930.
67. Cully M, You H, Levine AJ, et al. Beyond PTEN mutations: The PI3 K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192.
68. Castellvi J, Garcia A, Rojo F, et al. Phosphorylated 4E binding protein 1: A hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer. 2006;107:1801–1811.
69. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414.
70. LoPiccolo J, Blumenthal GM, Bernstein WB, et al. Targeting the PI3 K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50.
71. Marone R, Cmiljanovic V, Giese B, et al. Targeting phosphoinositide 3-kinase: Moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185.
72. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484.
73. Gadducci A, Tana R, Cosio S, et al. Molecular target therapies in endometrial cancer: From the basic research to the clinic. Gynecol Endocrinol. 2008;24:239–249.
74. Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456.
75. FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med. 2007;356:1915–1927.
76. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1–27.
77. Klein G, Klein E. Surveillance against tumors—is it mainly immunological? Immunol Lett. 2005;100:29–33.
78. Grulich AE, van Leeuwen MT, Falster MO, et al. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: A meta-analysis. Lancet. 2007; 370:59–67.
79. Owen M. T-cell receptors and MHC molecules. In: Roitt I, Borstoff J, Male D, eds. Immunology. 5th ed. St. Louis, MO: Mosby-Year Book; 1998.
80. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307.
81. Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238.
82. Bronte V. TH17 and cancer: Friends or foes? Blood. 2008;112:214.
83. Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213.
84. Hwang WT, Adams SF, Tahirovic E, et al. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol Oncol. 2012;124:192–198.
85. Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949.
86. Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–18543.
87. Sakaguchi S, Sakaguchi N, Shimizu J, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: Their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32.
88. Shevach EM. CD4+ CD25+ suppressor T cells: More questions than answers. Nat Rev Immunol. 2002;2:389–400.
89. Bookman MA, Bast RC Jr. The immunobiology and immunotherapy of ovarian cancer. Semin Oncol. 1991;18:270–291.
90. Bookman MA. Biological therapy of ovarian cancer: Current directions. Semin Oncol. 1998;25:381–396.
91. Mantia-Smaldone GM, Corr B, Chu CS. Immunotherapy in ovarian cancer. Hum Vaccin Immunother. 2012;8(9):1179–1191. doi:10.4161/hv.20738. Review.
92. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39(1):61–73.
93. Charbonneau B, Goode EL, Kalli KR, et al. The immune system in the pathogenesis of ovarian cancer. Crit Rev Immunol. 2013;33(2):137–164. Review.
94. Hopkins TG, Wood N. Female human papillomavirus (HPV) vaccination: Global uptake and the impact of attitudes. Vaccine. 2013;31(13):1673–1679.
95. Liu B, Nash J, Runowicz C, et al. Ovarian cancer immunotherapy: Opportunities, progresses and challenges. J Hematol Oncol. 2010;3:7. PMID:20146807.
96. Odunsi K, Jungbluth AA, Stockert E, et al. NY-ESO-1 and LAGE-1 cancer- testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 2003;63:6076–6083.
97. Odunsi K, Qian F, Matsuzaki J, et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci U S A. 2007;104:12837–12842.
98. Diefenbach CS, Gnjatic S, Sabbatini P, et al. Safety and immunogenicity study of NY-ESO-1b peptide and montanide ISA-51 vaccination of patients with epithelial ovarian cancer in high-risk first remission. Clin Cancer Res. 2008;14:2740–2748.
99. Odunsi KRK, Lele S, et al. Diversified prime and boost vaccination using recombinant vaccinia and fowlpox expressing NY-ESO-1 efficiently induces anti- body, CD4+ and CD8+ anti tumor immune responses in patients with ovarian cancer. Proceedings of the 38th Society of Gynecologic Oncologists, San Diego, CA, USA. Abstract 200.
100. Rahma OE, Ashtar E, Czystowska M, et al. A Gynecologic Oncology Group phase II trial of two p53 peptide vaccine approaches: Subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol Immunother. 2012;61:373–384.
101. Leffers N, Lambeck AJ, Gooden MJ, et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int J Cancer. 2009;125:2104–2113.
102. Leffers N, Vermeij R, Hoogeboom BN, et al. Long-term clinical and immunological effects of p53-SLP® vaccine in patients with ovarian cancer. Int J Cancer. 2012;130:105–112.
103. Morse MA, Secord AA, Blackwell K, et al. MHC class I-presented tumor antigens identified in ovarian cancer by immunoproteomic analysis are targets for T-cell responses against breast and ovarian cancer. Clin Cancer Res. 2011;17:3408–3419.
104. Gulley JL, Arlen PM, Tsang KY, et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. 2008;14:3060–3069.
105. Chiang CL, Kandalaft LE, Coukos G. Adjuvants for enhancing the immunogenicity of whole tumor cell vaccines. Int Rev Immunol. 2011;30:150–182.
106. Buckanovich RJ, Facciabene A, Kim S, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14:28–36.
107. Epenetos AA, Shepherd J, Britton KE, et al. 123I radioiodinated antibody imaging of occult ovarian cancer. Cancer. 1985;55:984–987.
108. Epenetos AA, Hooker G, Krausz T, et al. Antibody-guided irradiation of malignant ascites in ovarian cancer: A new therapeutic method possessing specificity against cancer cells. Obstet Gynecol. 1986;68:71S–74S.
109. Berek JS, Martinez-Maza O, Montz FJ. The immune system and gynecologic cancer. In: Coppelson MT, Morrow CP, eds. Gynecologic Oncology. Edinburgh: Churchill-Livingstone; 1992:119–151.
110. Ortiz-Sanchez E, Helguera G, Daniels TR, et al. Antibody-cytokine fusion proteins: Applications in cancer therapy. Expert Opin Biol Ther. 2008;8:609–632.
111. Kandalaft LE, Powell DJ Jr, Singh N, et al. Immunotherapy for ovarian cancer: What’s next? J Clin Oncol. 2011;29:925–933.
112. Berek JS, Taylor PT, Gordon A, et al. Randomized, placebo-controlled study of oregovomab for consolidation of clinical remission in patients with advanced ovarian cancer. J Clin Oncol. 2004; 22:3507–3516.
113. Berek JS, Taylor PT, Nicodemus CF. CA125 velocity at relapse is a highly significant predictor of survival post relapse: Results of a 5-year follow-up survey to a randomized placebo-controlled study of maintenance oregovomab immunotherapy in advanced ovarian cancer. J Immunother. 2008;31:207–214.
114. Berek J, Taylor P, McGuire W, et al. Oregovomab maintenance monoimmunotherapy does not improve outcomes in advanced ovarian cancer. J Clin Oncol. 2009;27:418–425.
115. Nicodemus CF, Chu C, Collins Y, et al. The immune adjuvant properties of front-line carboplatin-paclitaxel: A randomized phase 2 study of alternative schedules of intravenous oregovomab chemoimmunotherapy in advanced ovarian cancer. J Immunother. 2009;32:54–65.
116. Schlebusch H, Wagner U, Grunn U, et al. A monoclonal anti-idiotypic antibody ACA125 mimicking the tumor-associated antigen CA125 for immunotherapy of ovarian cancer. Hybridoma. 1995;14:167–174.
117. Wagner U. Antitumor antibodies for immunotherapy of ovarian carcinomas. Hybridoma. 1993;12:521–528.
118. Reinartz S, Köhler S, Schlebusch H, et al. Vaccination of patients with advanced ovarian carcinoma with the anti-idiotype ACA125: Immunological response and survival (phase Ib/II). Clin Cancer Res. 2004;10:1580–1587.
119. Wagner U, Köhler S, Reinartz S, et al. Immunological consolidation of ovarian carcinoma recurrences with monoclonal anti-idiotype antibody ACA125: Immune responses and survival in palliative treatment. Clin Cancer Res. 2001;7:1154–1162. Commentary by Foon K, Bhattacharya-Chatterjee M. Are solid tumor anti-idiotype vaccines ready for prime time? Clin Cancer Res. 2001;7:1112–1115.
120. Sabbatini P, Dupont J, Aghajanian C, et al. Phase I study of abagovomab in patients with epithelial ovarian, fallopian tube, or primary peritoneal cancer. Clin Cancer Res. 2006;12:5503–5510.
121. Rosenberg SA. Immunotherapy of cancer by systemic administration of lymphoid cells plus interleukin-2. J Biol Response Mod. 1984; 3:501–511.
122. Rosenberg SA, Lotze MT. Cancer immunotherapy using interleukin-2 and interleukin-2-activated lymphocytes. Annu Rev Immunol. 1986; 4:681–709.
123. Rosenberg SA, Lotze MT, Muul LM, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313:1485–1492.
124. Lotzova E. Role of human circulating and tumor-infiltrating lymphocytes in cancer defense and treatment. Nat Immun Cell Growth Regul. 1990;9:253–264.
125. Urba WJ, Clark JW, Steis RG, et al. Intraperitoneal lymphokine-activated killer cell/interleukin-2 therapy in patients with intra-abdominal cancer: Immunologic considerations. J Natl Cancer Inst. 1989;81:602–611.
126. Steis RG, Urba WJ, VanderMolen LA, et al. Intraperitoneal lymphokine-activated killer-cell and interleukin-2 therapy for malignancies limited to the peritoneal cavity. J Clin Oncol. 1990;8:1618–1629.
127. Berek JS, Lichtenstein AK, Knox RM, et al. Synergistic effects of combination sequential immunotherapies in a murine ovarian cancer model. Cancer Res. 1985;45:4215–4218.
128. West WH, Tauer KW, Yannelli JR, et al. Constant-infusion recombinant interleukin-2 in adoptive immunotherapy of advanced cancer. N Engl J Med. 1987;316:898–905.
129. Topalian SL, Solomon D, Avis FP, et al. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: A pilot study. J Clin Oncol. 1988;6:839–853.
130. Aoki Y, Takakuwa K, Kodama S, et al. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991;51:1934–1939.
131. Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following nonmyeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357.
132. Gotlieb WH, Abrams JS, Watson JM, et al. Presence of interleukin 10 (IL-10) in the ascites of patients with ovarian and other intra-abdominal cancers. Cytokine. 1992;4:385–390.
133. Disis ML, Schiffman K, Guthrie K, et al. Effect of dose on immune response in patients vaccinated with an HER-2/neu intracellular domain protein–based vaccine. J Clin Oncol. 2004;22:1916–1925.
134. Brulet JM, Maudoux F, Thomas S, et al. DNA vaccine encoding endosome-targeted human papillomavirus type 16 E7 protein generates CD4+ T cell-dependent protection. Eur J Immunol. 2007;37:376–384.
135. Wu TC, Guarnieri FG, Staveley-O’Carroll KF, et al. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci U S A. 1995;92:11671–11675.
136. Ji H, Wang TL, Chen CH, et al. Targeting human papillomavirus type 16 E7 to the endosomal/lysosomal compartment enhances the antitumor immunity of DNA vaccines against murine human papillomavirus type 16 E7-expressing tumors. Hum Gene Ther. 1999; 10:2727–2740.
137. Santin AD, Hermonat PL, Ravaggi A, et al. In vitro induction of tumor-specific human lymphocyte antigen class I-restricted CD8 cytotoxic T lymphocytes by ovarian tumor antigen-pulsed autologous dendritic cells from patients with advanced ovarian cancer. Am J Obstet Gynecol. 2000;183:601–609.
138. Santin AD, Hermonat PL, Ravaggi A, et al. Development, characterization and distribution of adoptively transferred peripheral blood lymphocytes primed by human papillomavirus 18 E7–pulsed autologous dendritic cells in a patient with metastatic adenocarcinoma of the uterine cervix. Eur J Gynaecol Oncol. 2000;21:17–23.
139. Zhao X, Wei YQ, Peng ZL. Induction of T cell responses against autologous ovarian tumors with whole tumor cell lysate-pulsed dendritic cells. Immunol Invest. 2001;30:33–45.
140. Hernando JJ, Park TW, Kubler K, et al. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: Clinical and immunological evaluation of a phase I trial. Cancer Immunol Immunother. 2002;51:45–52.
141. Inaba K, Turley S, Yamaide F, et al. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J Exp Med. 1998;188:2163–2173.
142. Mackensen A, Herbst B, Chen JL, et al. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoietic progenitor cells. Int J Cancer. 2000;86:385–392.
143. Thurner B, Roder C, Dieckmann D, et al. Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J Immunol Methods. 1999;223:1–15.
144. Thurner B, Haendle I, Roder C, et al. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190:1669–1678.
145. Finn OJ. Cancer vaccines: Between the idea and the reality. Nat Rev Immunol. 2003;3:630–641.
146. Paul S, Acres B, Limacher JM, et al. Cancer vaccines: Challenges and outlook in the field. IDrugs. 2007;10:324–328.
147. Tabi Z, Man S. Challenges for cancer vaccine development. Adv Drug Deliv Rev. 2006;58:902–915.
148. Cannon MJ, O’Brien TJ, Underwood LJ, et al. Novel target antigens for dendritic cell-based immunotherapy against ovarian cancer. Exp Rev Anticancer Ther. 2002;2:97–105.
149. McIlroy D, Gregoire M. Optimizing dendritic cell-based anticancer immunotherapy: Maturation state does have clinical impact. Cancer Immunol Immunother. 2003;52:583–591.
150. McIlroy D, Tanguy-Royer S, Le Meur N, et al. Profiling dendritic cell maturation with dedicated microarrays. J Leukoc Biol. 2005;78:794–803.
151. Berek JS, Knapp RC, Hacker NF, et al. Intraperitoneal immunotherapy of epithelial ovarian carcinoma with Corynebacterium parvum. Am J Obstet Gynecol. 1985;152:1003–1010.
152. Lichtenstein A, Berek J, Bast R, et al. Activation of peritoneal lymphocyte cytotoxicity in patients with ovarian cancer by intraperitoneal treatment with Corynebacterium parvum. J Biol Response Mod. 1984;3:371–378.
153. Bast RC Jr, Berek JS, Obrist R, et al. Intraperitoneal immunotherapy of human ovarian carcinoma with Corynebacterium parvum. Cancer Res. 1983;43:1395–1401.
154. Mantovani A, Sessa C, Peri G, et al. Intraperitoneal administration of Corynebacterium parvum in patients with ascitic ovarian tumors resistant to chemotherapy: Effects on cytotoxicity of tumor-associated macrophages and NK cells. Int J Cancer. 1981;27:437–446.
155. Gusdon JP Jr, Homesley HD, Jobson VW, et al. Treatment of advanced ovarian malignancy with chemoimmunotherapy using autologous tumor and Corynebacterium parvum. Obstet Gynecol. 1983; 62:728–735.
156. Howell SB, Kirmani S, Lucas WE, et al. A phase II trial of intraperitoneal cisplatin and etoposide for primary treatment of ovarian epithelial cancer. J Clin Oncol. 1990;8:137–145.
157. Edwards RP, Gooding W, Lembersky BC, et al. Comparison of toxicity and survival following intraperitoneal recombinant interleukin-2 for persistent ovarian cancer after platinum: Twenty-four-hour versus 7-day infusion. J Clin Oncol. 1997;15:3399–3407.
158. Vlad AM, Budiu RA, Lenzner DE, et al. A phase II trial of intraperitoneal interleukin-2 in patients with platinum-resistant or platinum-refractory ovarian cancer. Cancer Immunol Immunother. 2010;59:293–301.
159. Nardi M, Cognetti F, Pollera CF, et al. Intraperitoneal recombinant alpha-2-interferon alternating with cisplatin as salvage therapy for minimal residual-disease ovarian cancer: A phase II study. J Clin Oncol. 1990;8:1036–1041.
160. Repetto L, Chiara S, Guido T, et al. Intraperitoneal chemotherapy with carboplatin and interferon alpha in the treatment of relapsed ovarian cancer: A pilot study. Anticancer Res. 1991;11:1641–1643.
161. Hurteau JA, Blessing JA, DeCesare SL, et al. Evaluation of recombinant human interleukin-12 in patients with recurrent or refractory ovarian cancer: A Gynecologic Oncology Group study. Gynecol Oncol. 2001;82:7–10.
162. Lenzi R, Rosenblum M, Verschraegen C, et al. Phase I study of intraperitoneal recombinant human interleukin 12 in patients with Müllerian carcinoma, gastrointestinal primary malignancies, and mesothelioma. Clin Cancer Res. 2002;8:3686–3695.
163. Recchia F, Saggio G, Cesta A, et al. Interleukin-2 and 13-cis retinoic acid as maintenance therapy in advanced ovarian cancer. Int J Oncol. 2005;27:1039–1046.
164. Roche MR, Rudd PJ, Krasner CN, et al. Phase II trial of GM-CSF in women with asymptomatic recurrent müllerian tumors. Gynecol Oncol. 2010;116:168–172.
165. Schmeler KM, Vadhan-Raj S, Ramirez PT, et al. A phase II study of GM-CSF and rIFN-gamma1b plus carboplatin for the treatment of recurrent, platinum-sensitive ovarian, fallopian tube and primary peritoneal cancer. Gynecol Oncol. 2009;113:210–215.
166. Woo EY, Chu CS, Goletz TJ, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–4772.
167. Powell DJ Jr, Felipe-Silva A, Merino MJ, et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J Immunol. 2007;179:4919–4928.
168. Kryczek I, Wei S, Zou L, et al. Cutting edge: TH17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J Immunol. 2007;178:6730–6733.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


