Cancer is a complex disease that arises because of genetic and epigenetic alterations that disrupt cellular proliferation, senescence, and death (Fig. 1.1). The alterations that underlie the development of cancers have a diverse etiology, and the loss of DNA repair mechanisms often plays a role in allowing mutations to accumulate. Specific molecular changes that cause a normal cell to become malignant have been identified, but their spectrum varies considerably between cancer types.
The malignant phenotype is characterized by the ability to invade surrounding tissues and metastasize. The development of a cancer elicits a considerable molecular response in the local microenvironment that is characterized by recruitment of stromal elements such as new blood vessels and by an active immunologic response. These secondary events play a critical role in the evolution and progression of cancers. Although the molecular pathogenesis of gynecologic cancers has been only partially elucidated, advances in the understanding of these diseases are providing the opportunity for improvements in diagnosis, treatment, and prevention.
The initial sections of this chapter will outline what is known regarding the basic molecular mechanisms involved in the development of cancers and the evolution of the malignant phenotype. The molecular alterations characteristic of gynecologic cancers will be outlined in the later sections.
Growth Regulation
Proliferation
The number of cells in normal tissues is tightly regulated by a balance between cellular proliferation and death. The final common pathway for cell division involves distinct molecular switches that control cell cycle progression from G1 to the S phase of DNA synthesis. These include the retinoblastoma (Rb) and E2F proteins and their various regulatory cyclins, cyclin-dependent kinases (cdks), and cdk inhibitors. Likewise, the events that facilitate progression from G2 to mitosis and cell division are regulated by other cyclins and cdks (Fig. 1.2).
In some tissues—such as the bone marrow, epidermis and gastrointestinal tract—the life span of mature cells is relatively short, and high rates of proliferation by progenitor cells are required to maintain the population. In other tissues—such as liver, muscle, and brain—cells are long lived, and proliferation rarely occurs. Complex molecular mechanisms have evolved to closely regulate proliferation. These involve a finely tuned balance between stimulatory and inhibitory growth signals.
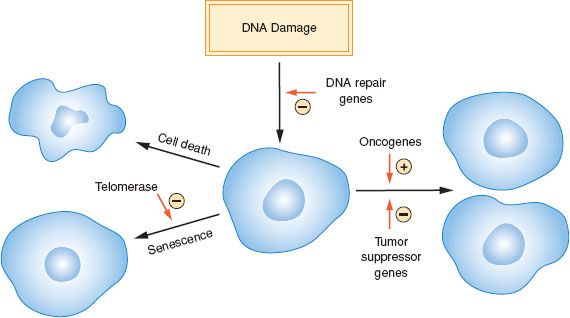
Figure 1.1 Role of proliferation, cell death, senescence, and DNA damage in cancer development.
Dysregulation of cellular proliferation is one of the main hallmarks of cancer. There may be increased activity of genes involved in stimulating proliferation (oncogenes) or loss of growth inhibitory (tumor suppressor) genes or both. In the past, it was thought that cancer might arise solely because of more rapid proliferation or a higher fraction of proliferating cells. Although increased proliferation is a characteristic of many cancers and is an appealing therapeutic target (1), the fraction of cancer cells actively dividing, and the time required to transit the cell cycle, is not strikingly different between many cancers and corresponding normal cells of the same lineage. Altered regulation of proliferation is only one of several factors that contribute to malignant transformation. Ultimately, the relative balance of growth stimulatory factors and inhibitory factors is shifted in malignancy to promote unregulated cellular proliferation.
Cell Death
In addition to being driven by increased proliferation, growth of a cancer may be attributable to cellular resistance to death. At least three distinct types of cell death pathways have been characterized, including apoptosis, necrosis, and autophagy (2). All three pathways may be ongoing simultaneously within a tumor and methods that distinguish between them are far from perfect.
Apoptosis
The term apoptosis is derived from Greek, and alludes to a process akin to leaves dying and falling off a tree. Apoptosis is an active, energy-dependent process that involves cleavage of the DNA by endonucleases and proteins by proteases called caspases. Morphologically, apoptosis is characterized by condensation of chromatin, nuclear and cytoplasmic blebbing, and cellular shrinkage. The molecular events that affect apoptosis in response to various stimuli are complex and have been only partially elucidated (3), but several reliable markers of apoptosis have been discovered including annexin V, caspase-3 activation, and DNA fragmentation (4).
External stimuli such as tumor necrosis factor, tumor necrosis factor-related apoptosis-inducing ligand, fatty acid synthase (Fas), and other death ligands that interact with cell surface receptors can induce activation of caspases, and lead to apoptosis via an extrinsic pathway (Fig. 1.3). The intrinsic pathway is activated in response to a wide range of stresses including DNA damage and deprivation of growth factors. The intrinsic apoptosis pathway is regulated by a complex interaction of proapoptotic and antiapoptotic proteins in the mitochondrial membrane that affect its permeability. Proteins that increase permeability allow the release of cytochrome c, which activates the apoptosome complex leading to the activation of caspases leading to apoptosis. Conversely, proteins that stabilize mitochondrial membranes inhibit apoptosis. The first major insight that led to the understanding of the intrinsic apoptotic pathway was the finding that an activating translocation of the bcl-2 gene in B-cell lymphomas resulted in essentially a complete inhibition of apoptosis (5). Subsequent studies have demonstrated that the antiapoptotic effect of bcl-2 is attributable to stabilization of the mitochondrial membrane. Additional genes related to bcl-2 (such as BAD, BCL-XL, and others) block apoptosis by inhibiting membrane permeability. Other genes in the BCL family (such as BAX, BAK, and others) increase membrane permeability and are proapoptotic. An increased understanding of the complex system of molecular checks and balances involved in regulation of apoptosis provides opportunities for targeted cancer therapies; several strategies are under development (6).
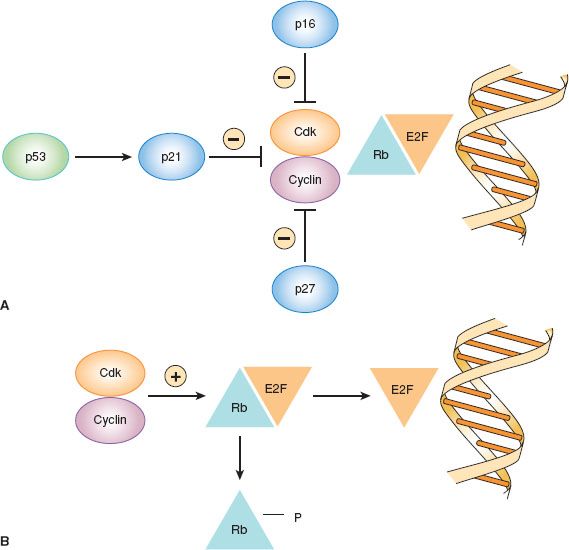
Figure 1.2 Regulation of cell cycle arrest in G1 by cyclin-dependent kinase (cdk) inhibitors. A: Cell cycle arrest. B: Cell cycle progression.
In addition to restraining the number of cells in a population, apoptosis serves an important role in preventing malignant transformation by allowing the elimination of cells that have undergone genetic damage. Following exposure of cells to mutagenic stimuli, including radiation and carcinogenic drugs, the cell cycle is arrested so that DNA damage may be repaired. If DNA repair is not sufficient, apoptosis occurs so that damaged cells do not survive. This serves as an anticancer surveillance mechanism by which mutated cells are eliminated before they become fully transformed. In this regard, the TP53 tumor suppressor gene is a critical regulator of cell cycle arrest and apoptosis in response to DNA damage, and the frequency of TP53 mutations in human cancers reflects its critical role in preventing tumorigenesis.

Figure 1.3 Apoptosis pathways.
Necrosis
Necrosis is a type of cell death that is distinct from apoptosis, and is the result of bioenergetic compromise (4). Morphologic changes include swollen organelles and rupture of the cell membrane, leading to loss of osmoregulation and cellular fragmentation. Necrosis is a less well-regulated process that leads to spillage of protein contents, and this may incite a brisk immune response. This is in contrast to the silent elimination of cells by apoptosis, which typically elicits a minimal immune response. There is evidence that some drugs may enhance necrotic death in tumors, and this may stimulate a beneficial antitumor immune response (4).
Autophagy
Autophagy is a potentially reversible process in which a cell that is stressed “eats” itself (4). A wide range of stresses have been identified that may elicit autophagy (some of which may also elicit apoptosis), including growth factor deprivation and accumulation of reactive oxygen species. Unlike necrosis and apoptosis—in which the loss of integrity of the cytoplasmic and nuclear membranes, respectively, are defining events—autophagy is characterized by the formation of cytoplasmic autophagic vesicles, into which cellular proteins and organelles are sequestered. This may allow for cell survival if damaged organelles can be repaired. Conversely, the process may lead to cell death if these vesicles fuse with lysosomes with resultant degradation of their contents. Several cancer therapeutic agents have been shown to induce autophagy, while targeted disruption of genes such as ATG5 that are involved in autophagy can inhibit cell death (4).
Cellular Senescence
Normal cells are capable of undergoing division only a finite number of times before becoming senescent. Cellular senescence is regulated by a biologic clock related to progressive shortening of repetitive DNA sequences (TTAGGG) called telomeres that cap the ends of each chromosome. Telomeres are thought to be involved in chromosomal stabilization and in preventing recombination during mitosis. At birth, chromosomes have long telomeric sequences (150,000 bases) that become progressively shorter by 50 to 200 bases each time a cell divides. Telomeric shortening is the molecular clock that triggers senescence. Malignant cells often avoid senescence by turning on expression of telomerase activity to prevent telomeric shortening (7). Telomerase is a ribonucleoprotein complex, and both the protein and RNA subunits have been identified. The RNA component serves as a template for telomeric extension, and the protein subunit catalyzes the synthesis of new telomeric repeats.
Telomerase activity is detectable in a high fraction of many cancers, including ovarian (8), cervical (9,10), and endometrial (11). It has been suggested that detection of telomerase might be useful for early diagnosis of cancer, but the lack of specificity is a significant issue. In this regard, endometrium is one of the normal adult tissues in which telomerase expression is most common (12). Perhaps this relates to the need for a large number of lifetime cell divisions because of the rapid growth and shedding of this tissue each month during the reproductive years. Therapeutic approaches to inhibiting telomerase are under development, focusing on reversing the immortalized state of cancer cells to make them susceptible once again to normal replicative senescence (7).
Origins of Genetic Alterations
Human cancers arise because of a series of genetic and epigenetic alterations that lead to disruption of normal mechanisms that govern cell growth, death and senescence (13,14). Genetic damage may be inherited or may arise after birth as a result of either exposure to exogenous carcinogens or endogenous mutagenic processes within the cell (Table 1.1). The incidence of most cancers increases with aging because the longer one is alive, the higher the likelihood that a cell will acquire sufficient damage to become fully transformed. It is thought that at least three to six critical “driver” alterations are required to fully transform a cell. However, most cancer cells are genetically unstable, with an average of 30 to 100 acquired mutations per cancer. Some of these may be simply “passenger” mutations that occur as a result of generalized genetic instability. Although not involved in malignant transformation, these may contribute to evolution of the malignant phenotype with respect to growth, invasion, metastasis, and response to therapy, among other characteristics. Genetic instability also results in evolution of heterogeneous clones within a tumor. There is evidence that small numbers of progenitor cells (stem cells) exist within a tumor and have the capacity to regenerate tumors. The stem cell theory suggests that these dormant or quiescent stem cells may be more resistant to therapy, and thus responsible for the development of recurrent disease (15).
Table 1.1 Origins of Genetic Damage in Human Cancers

Inherited Cancer Susceptibility
Although most cancers arise sporadically in the population because of acquired genetic damage, inherited mutations in cancer susceptibility genes are responsible for some cases. Families with these mutations exhibit a high incidence of specific types of cancers. The age of cancer onset is younger in these families and it is not unusual for some individuals to be affected with multiple primary cancers.
The most common forms of hereditary cancer syndromes predispose to breast/ovarian (BRCA1, BRCA2) and colon/endometrial (Lynch syndrome genes such as MSH2 and MLH1) cancers (Table 1.2). Examples of other hereditary cancer syndromes are outlined in Table 1.2. Beyond this list of known hereditary cancer syndromes, additional high penetrance genes have been discovered that are mutated infrequently but confer dramatically increased cancer risks. For example, in addition to BRCA1–2, germline mutations in a number of other genes in the homologous recombination (HR) DNA repair pathway confer susceptibility to breast/ovarian cancer (e.g., RAD51 C/D, BRIP1, PALB2). This is leading to the development of hereditary cancer genetic test panels that will likely replace testing for individual genes.
Table 1.2 Hereditary Cancer Syndromes
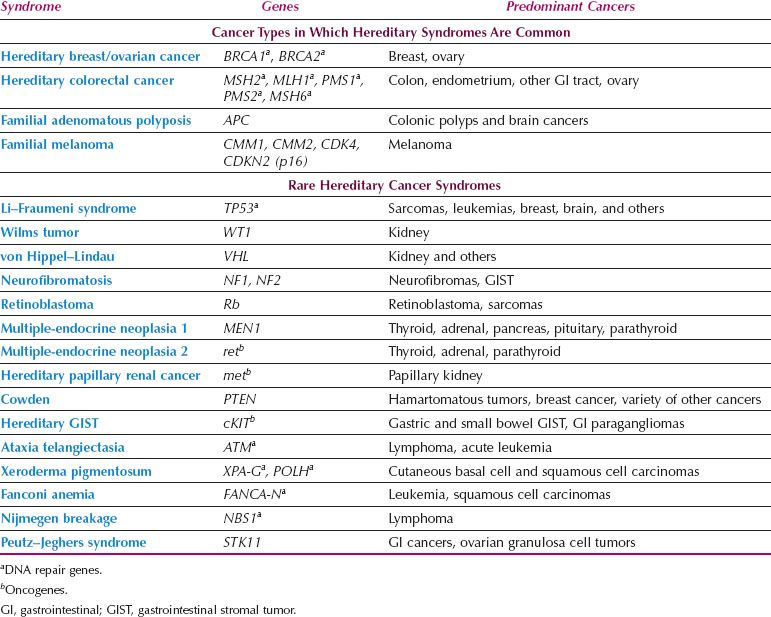
Tumor suppressor genes have been implicated most frequently in hereditary cancer syndromes, and many of these are DNA repair genes such as BRCA1–2 and the Lynch syndrome genes. In only a few instances are germline mutations in oncogenes responsible for hereditary cancers (Table 1.2). Although affected individuals carry the germline alteration in every cell of their bodies, paradoxically, cancer susceptibility genes are characterized by a limited repertoire of cancers. There is no relationship between expression patterns of these genes in various organs and the development of specific types of cancers. For example, BRCA1 expression is high in the testis, but men who inherit mutations in this gene are not predisposed to develop testicular cancer. The penetrance of cancer susceptibility genes is incomplete because not all individuals who inherit a mutation develop cancer. The emergence of cancers in carriers depends on the occurrence of additional genetic alterations.
The familial cancer syndromes described above result from rare mutations that occur in less than 1% of the population. Low-penetrance common genetic polymorphisms may also affect cancer susceptibility, albeit less dramatically (16). There are more than 10 million polymorphic genetic loci in the human genome, and many of these polymorphisms are common in the population. Although genetic polymorphisms do not increase risk sufficiently to produce familial cancer clustering, they could account for a significant fraction of cancers currently classified as sporadic, because of their relatively high prevalence. For example, 6% of Ashkenazi Jews carry the less common allele of a polymorphism in codon 1,307 of the APC gene that changes a single amino acid in the protein, and this increases the risk of colorectal cancer by about 50% (17). The recent development of genomic technologies that can assess hundreds of thousands of polymorphisms simultaneously in large numbers of individuals is fueling the search for additional genetic susceptibility polymorphisms (16). It is estimated that dozens to a few hundred of these likely exist for each cancer type. A more complete understanding of the genetic factors that affect cancer susceptibility could facilitate implementation of screening and preventive approaches in subsets of the population at increased risk.
Acquired Genetic Damage
The etiology of acquired genetic damage in cancers also has been elucidated to some extent. For example, a strong causal link exists between cigarette smoke and cancers of the aerodigestive tract and between ultraviolet radiation and skin cancer. For many common forms of cancer (colon, breast, endometrium, ovary), a strong association with specific carcinogens does not exist. It is thought that the genetic alterations responsible for these cancers arise mainly because of endogenous mutagenic processes such as methylation, deamination, and hydrolysis of DNA. Furthermore, spontaneous errors in DNA synthesis may occur during the process of DNA replication associated with normal proliferation. Finally, free radicals generated in response to inflammation and other cellular damage may cause DNA damage. These endogenous processes produce many mutations each day in every cell in the body. While the multiple cellular mechanisms for DNA damage surveillance and repair are highly effective, some mutations may elude them. The efficiency of these DNA damage-response systems varies between individuals because of genetic and other factors and may affect susceptibility to cancer.
Epigenetic Changes
Epigenetics changes are heritable changes that do not result from alterations in DNA sequence (14). Methylation of cytosine residues that reside next to guanine residues is the primary mechanism of epigenetic regulation, and this process is regulated by a family of DNA methyltransferases. Most cancers have globally reduced DNA methylation, which may contribute to genomic instability. Conversely, selective hypermethylation of cytosines in the promoter regions of tumor suppressor genes may lead to their inactivation and contribute to carcinogenesis.
There is a family of imprinted genes in which either the maternal or paternal copy is normally completely silenced because of methylation. Loss of imprinting in genes that stimulate proliferation, such as insulin-like growth factor 2 (IGF2), may provide an oncogenic stimulus further disrupting the balance between proliferation and cell death. Acetylation and methylation of the histone proteins that coat DNA represent another level of epigenetic regulation that is altered in cancer. While the underlying cause of these epigenetic alterations remains poorly understood, they represent appealing therapeutic targets. Histone deacetylase inhibitors and hypomethylating agents can reactivate the expression of genes silenced by acetylation and methylation, respectively, and are used for the treatment of some hematologic malignancies, such as myelodysplastic syndromes and lymphomas.
Oncogenes
Alterations in genes that stimulate cellular growth (oncogenes) can cause malignant transformation (13). Oncogenes can be activated via several mechanisms. Amplification of some oncogenes results in multiple copies of a gene with resultant overexpression of the corresponding protein. HER2-neu amplification in a subset of breast cancers is an example of a tumor driven by amplification of a single gene that can be therapeutically targeted using an anti-HER2 antibody, trastuzumab. Other oncogenes, such as KIT in gastrointestinal stromal tumors (GIST), may become overactive when affected by point mutations at codons that change a single amino acid leading to gain of function. Finally, oncogenes may be translocated from one chromosomal location to another and then come under the influence of promoter sequences that cause overexpression of the gene. This latter mechanism frequently occurs in leukemias and lymphomas (e.g., the BCR-ABL translocation in chronic myelogenous leukemia), but is relatively uncommon in gynecologic and other solid tumors. For tumorigenesis driven by activation of individual oncogenes, targeting the oncogene can be a useful therapeutic approach.
In cell culture systems, many genes that are involved in normal growth regulatory pathways can elicit transformation when altered to overactive forms via amplification, mutation, or translocation. On this basis, a large number of genes have been classified as oncogenes. Studies in human cancers have suggested that the actual spectrum of genes altered in the development of human cancers is more limited. A number of genes that elicit transformation when activated in vitro have not been documented to undergo alterations in human cancers. In this section, the various classes of oncogenes will be summarized and particular attention paid to those that are altered in gynecologic cancers.
Cell Membrane Oncogenes—Peptide Growth Factors and Their Receptors
Peptide growth factors in the extracellular space–such as those of the epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF) families—stimulate a cascade of molecular events that leads to proliferation by binding to cell membrane receptors. Growth factors are involved in normal cellular processes such as development, stromal–epithelial communication, tissue regeneration, and wound healing. Unlike endocrine hormones, which are secreted into the blood stream and act in distant target organs, peptide growth factors typically act in the local environment where they have been secreted.
Aberrant proliferative signaling through these peptide growth factor pathways can occur through a number of mechanisms: (1) Increased or inappropriate autocrine production of growth factors, (2) increased paracrine production of growth factors by tumor stromal environment, (3) increased responsiveness of receptors to growth factor ligands or ligand independent activation of the receptor, and (4) constitutive activation of components downstream of the receptor. Autocrine growth stimulation may be a key strategy by which cancer cell proliferation becomes autonomous. In this model, it is postulated that cancers secrete stimulatory growth factors that interact with receptors on the same cell. Although peptide growth factors provide a growth stimulatory signal, there is little evidence to suggest that overproduction of growth factors is a precipitating event in the development of most cancers. Increased expression of peptide growth factors likely serves to promote rather than initiate malignant transformation.
Cell membrane receptors that bind peptide growth factors are composed of an extracellular ligand-binding domain, a membrane spanning region, and a cytoplasmic tyrosine kinase domain (18). Binding of a growth factor to the extracellular domain results in aggregation and conformational shifts in the receptor and activation of the inner tyrosine kinase (Fig. 1.4). This kinase phosphorylates tyrosine residues on both the growth factor receptor itself (autophosphorylation) and on molecular targets in the cell interior, leading to activation of secondary signals.
Growth of some cancers is driven by overexpression of receptor tyrosine kinases. Therapeutic strategies that target receptor tyrosine kinases have been an active area of investigation. Trastuzumab is a monoclonal antibody that blocks the HER-2/neu receptor and it is widely used in the treatment of breast and gastric cancers that overexpress this tyrosine kinase (19). Cetuximab is a monoclonal antibody that targets the epidermal growth factor receptor (EGFR), whereas gefitinib is a direct inhibitor of the EGFR tyrosine kinase (20). Both are used in the treatment of EGFR-expressing nonsmall cell lung cancers. Lapatinib is a dual EGFR/HER-2 kinase inhibitor approved for treatment of HER-2 overexpressing breast cancers. Imatinib antagonizes the activity of the BCR-ABL, c-kit, and PDGF receptor tyrosine kinases and has proven effective in treatment of chronic myelogenous leukemias and gastrointestinal stromal tumors.

Figure 1.4 Mitogenic signal transduction pathways.
WNT Pathway
β-catenin (CTNNB1) is involved along with cadherins in cell-cell adhesion junctions and may play a role in inhibition of excessive growth when cells come in contact with each other (Fig. 1.5). β-catenin may be translocated to the nucleus and play a role in regulating transcription of genes involved in embryonic development, cell differentiation and cell polarity. β-catenin activity is regulated by the WNT pathway (21,22). The WNT family of genes encodes secreted peptides that interact with Frizzled family cell surface receptors. Frizzled activates the intracellular Dishevelled protein, resulting in an increase in the amount of β-catenin translocated to the nucleus. Dishevelled accomplishes this by inhibiting a complex of proteins that includes axin, GSK-3β, and APC that normally promote proteolytic degradation of β-catenin. This allows β-catenin to enter the nucleus and interact with TCF/LEF family transcription factors to promote gene expression.
Genes encoding WNT signaling inhibitors are often downregulated during carcinogenesis and driver mutations in several of these genes (APC, Axin, GSK-3β, β-catenin) occur frequently in human cancers, including endometrial cancers. Germline mutations in the APC gene are responsible for familial adenomatous polyposis of the colon.
Intracellular Oncogenes
Following the interaction of peptide growth factors and their receptors, secondary molecular signals are generated to transmit the growth stimulus to the nucleus. This function is served by a multitude of complex and overlapping signal transduction pathways that occur in the inner cell membrane and cytoplasm. Many of these signals involve phosphorylation of proteins by enzymes known as nonreceptor kinases (23). These kinases transfer a phosphate group from ATP to specific amino acid residues of target proteins. The kinases that are involved in growth regulation include those that phosphorylate tyrosine residues on proteins, and others that are specific for serine or threonine residues such as AKT (24). In addition, phosphatidylinositol (PIK) 3-kinases are a class of growth regulatory lipid kinases that phosphorylate inositol in the cell membrane (25). The activity of kinases is regulated by phosphatases, such as PTEN, which act in opposition to the kinases by removing phosphates from the target proteins (Fig. 1.6).
Guanosine-triphosphate–binding proteins (G proteins) represent another class of molecules involved in transmission of growth signals. They are located on the inner aspect of the cell membrane and have intrinsic GTPase activity that catalyzes the exchange of guanine-triphosphate (GTP) for guanine-diphosphate (GDP). In their active GTP-bound form, G proteins interact with kinases that are involved in relaying the mitogenic signal, such as those of the MAP kinase family. Conversely, hydrolysis of GTP to GDP, which is stimulated by GTPase-activating proteins (GAPs), leads to inactivation of G proteins.

Figure 1.5 Wingless (WNT) Beta-catenin signaling. WNT extracellular ligands bind Frizzled receptors and regulate the phosphorylation status of axin. Axin functions as part of the destruction complex that regulates the stability of Beta-catenin, a transcriptional regulator. (Redrawn from Berchuck A, Levine DA, Farley JH, et al. Molecular pathogenesis of gynecologic cancers. In: Barakat RR, ed. Principles and Practice of Gynecologic Oncology. Lippincott Williams and Wilkins; 2013.)
The ras family of G proteins is among the most frequently mutated oncogenes in human cancers (e.g., gastrointestinal and endometrial cancers). Activation of ras genes usually involves point mutations in codons 12, 13, or 61 that result in constitutively activated molecules (26). Therapeutic approaches to interfering with ras signaling are being developed. The most successful approach involves inhibition of BRAF, a kinase that interacts with ras proteins in activating the MAP kinase pathway. BRAF mutations occur in many cancers that lack ras mutations, such as melanoma, where a mutation resulting in the substitution of glutamic acid for valine at amino acid 600 (V600E) is common. Vemurafenib, an inhibitor of the kinase domain of mutant BRAF, improves survival of melanoma patients whose tumors carry a BRAF mutation; however, the tumors ultimately develop resistance to the BRAF inhibitor by developing other mechanisms to activate growth pathways and circumvent BRAF inhibition (27).
Nuclear Oncogenes
If proliferation is to occur in response to signals generated in the cell membrane and cytoplasm, these events must lead to activation of nuclear transcription factors and other genetic products responsible for stimulating DNA replication and cell division. Expression of several genes that encode nuclear proteins increases dramatically within minutes of treatment of cells with peptide growth factors. When induced, the products of these genes bind to specific DNA regulatory elements and induce transcription of genes involved in DNA synthesis and cell division. Examples include the fos and jun oncogenes, which dimerize to form the activator protein 1 (AP1) transcription complex. When inappropriately overexpressed, these transcription factors can act as oncogenes. Among the nuclear transcription factors involved in stimulating proliferation, amplification or overexpression of members of the myc family has most often been implicated in the development of human cancers. Many of the nuclear regulatory genes such as myc that control proliferation also affect the threshold for apoptosis. Genes that positively regulate cell cycle progression may be amplified and/or overexpressed in some cancers leading to unrestrained proliferation (e.g., cyclin D1, cyclin E1). Finally, as discussed previously, genes encoding nuclear proteins that inhibit apoptosis (e.g., bcl-2) can act as oncogenes when altered to constitutively active forms.

Figure 1.6 The phosphatidylinositol 3-kinase (PI3 K) pathway is activated by RAS and by a number of growth factor receptors, here exemplified by IGFIR and the RebB1/ErbB2 heterodimer. Activated PI3K generates phosphatidylinositol-3,4,5-triphophate (PIP3), which activates phosphoinositide-dependent kinse-1 (PDK). In turn, PDK phosphorylates AKT. PTEN is an endogenous inhibitor of AKT activation. Phosphorylated AKT tranduces multiple downstream signals, including activation of the mTOR and inhibition of the FOXO family of transcription factors. mTOR activation promotes the synthesis of proteins required for cell growth and cell cycle progression. (Redrawn from Berchuck A, Levine DA, Farley JH, et al. Molecular pathogenesis of gynecologic cancers. In: Barakat RR, ed. Principles and Practice of Gynecologic Oncology. Lippincott Williams and Wilkins; 2013.)
Tumor Suppressor Genes
Loss of tumor suppressor gene function plays a role in the development of most cancers. This usually involves a two-step process in which both copies of a tumor suppressor gene are inactivated. In most cases, there is mutation of one copy of a tumor suppressor gene and loss of the other copy caused by deletion of a segment of the chromosome where the gene resides. Some tumor suppressor genes may be inactivated because of methylation of the promoter region of the gene (14). The promoter is an area proximal to the coding sequence that regulates whether the gene is transcribed from DNA to RNA. When the promoter is methylated, it is resistant to activation and the gene is essentially silenced despite remaining structurally intact.
This two-hit paradigm is relevant to both hereditary cancer syndromes, in which one mutation is inherited and the second acquired, and sporadic cancers, in which the two hits are acquired. Tumor suppressor gene products are found throughout the cell, reflecting their diverse functions. With the recognition that inactivation of tumor suppressor genes is a defining feature of cancers, genetic therapeutic strategies have been developed that aim to deliver functional copies of these genes lost to cancer cells.
Nuclear Tumor Suppressor Genes
The retinoblastoma gene was the first tumor suppressor gene discovered (Table 1.2) (28). The Rb gene plays a key role in the regulation of cell cycle progression (Fig. 1.2). In the G1 phase of the cell cycle, Rb protein binds to the E2F transcription factor and prevents it from activating transcription of other genes involved in cell cycle progression. G1 arrest is maintained by cdk inhibitors that prevent phosphorylation of Rb, such as p16, p21, and p27 (29). When Rb is phosphorylated by cyclin–cdk complexes, E2F is released and stimulates entry into the DNA synthesis phase of the cell cycle. Other cyclins and cdks are involved in progression from G2 to mitosis. Mutations in the Rb gene have been noted primarily in retinoblastomas and sarcomas, but may occur rarely in other types of cancers. By maintaining G1 arrest, the cdk inhibitors p16, p21, p27, and others act as tumor suppressor genes. Loss of p16 tumor suppressor function as a result of genomic deletion or promoter methylation occurs in some cancers, including familial melanomas. Likewise, loss of p21 and p27 has been noted in some cancers.
Mutation of the TP53 tumor suppressor gene is the most frequent genetic event described in human cancers (Fig. 1.7) (30,31). The TP53 gene encodes a 393 amino acid protein that plays a central role in the regulation of both proliferation and apoptosis. In normal cells, p53 protein resides in the nucleus and exerts its tumor suppressor activity by binding to transcriptional regulatory elements of genes, such as the cdk inhibitor p21, that act to arrest cells in G1. The MDM2 gene product degrades p53 protein when appropriate, whereas p14ARF downregulates MDM2 when upregulation of p53 is needed to initiate cell cycle arrest.
Many cancers have missense mutations in one copy of the TP53 gene that result in substitution of a single amino acid in exons 5 through 8, which encode the DNA binding domains. Although these mutant TP53 genes encode full-length proteins, they are unable to bind to DNA and regulate transcription of other genes. Mutation of one copy of the TP53 gene often is accompanied by deletion of the other copy, leaving the cancer cell with only mutant p53 protein. If the cancer cell retains one normal copy of the TP53 gene, mutant p53 protein can complex with wild-type p53 protein and prevent it from oligimerizing and interacting with DNA. Because inactivation of both TP53 alleles is not required for loss of p53 function, mutant p53 is said to act in a “dominant negative” fashion. Although normal cells have low levels of p53 protein because it is rapidly degraded, missense mutations encode protein products that are resistant to degradation. The resultant overaccumulation of mutant p53 protein in the nucleus can be detected immunohistochemically. A smaller fraction of cancers have mutations in the TP53 gene that encode truncated protein products. In these cases, loss of the other allele occurs as the second event as is seen with other tumor suppressor genes.
Beyond simply inhibiting proliferation, normal p53 is thought to play a role in preventing cancer by stimulating apoptosis of cells that have undergone excessive genetic damage. In this regard, p53 has been described as the “guardian of the genome” because it delays entry into S phase until the genome has been cleansed of mutations. If DNA repair is inadequate, then p53 may initiate apoptosis, thereby eliminating cells with genetic damage. Likewise, other genes that repair damage to the DNA nucleotide sequence or strand breakage sometimes are classified as tumor suppressors. These other genes will be discussed in the next sections in the context of hereditary gynecologic cancer syndromes.
Extranuclear Tumor Suppressor Genes
Although many tumor suppressor genes—including TP53, Rb, and p16—encode nuclear proteins, some extranuclear tumor suppressors have been identified. Theoretically, any protein that normally is involved in inhibition of proliferation has the potential to act as a tumor suppressor. In this regard, appealing candidates include phosphatases such as PTEN that normally oppose the action of the tyrosine kinases by dephosphorylating tyrosine residues. In addition to its phosphatase activity, PTEN is homologous to the cytoskeleton proteins tensin and axin. It has been postulated that PTEN might act to inhibit invasion and metastasis through modulation of the cytoskeleton. The APC tumor suppressor gene encodes a cytoplasmic protein involved in the WNT signaling pathway that regulates both cellular proliferation and adhesion (see section on Adhesion). Inactivation of APC leads to malignant transformation and inherited mutations in this gene are responsible for familial adenomatous polyposis syndrome.
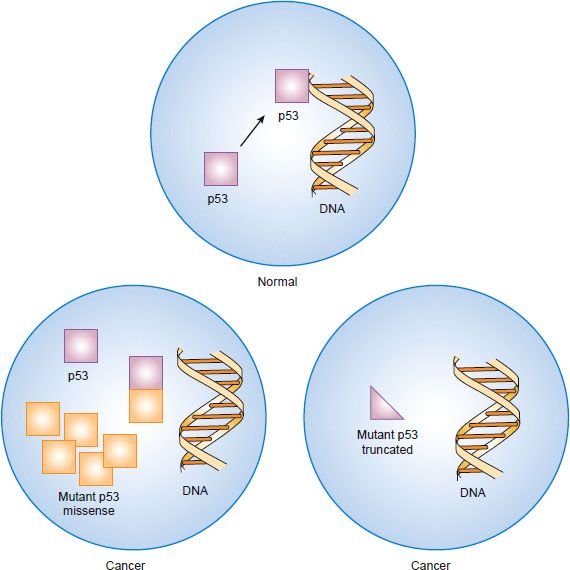
Figure 1.7 Inactivation of the p53 tumor suppressor gene by “dominant negative” missense mutation or by truncation mutation and deletion.
The transforming growth factor-beta (TGF-β) family of peptide growth factors inhibits proliferation of normal epithelial cells and serves as a tumor suppressive pathway (22). It is thought that TGF-β causes G1 arrest by inducing expression of cdk inhibitors such as p27. Three closely related forms of TGF-β that are encoded by separate genes (TGF-β1, TGF-β2, and TGF-β3) have been discovered. TGF-β is secreted from cells in an inactive form bound to a portion of its precursor molecule from which it must be cleaved to release biologically active TGF-β. Active TGF-β interacts with type I and type II cell surface TGF-β receptors and initiates serine or threonine kinase activity. Prominent intracellular targets include a class of molecules called Smads that translocate to the nucleus and act as transcriptional regulators.
MicroRNA
In addition to primary dysregulation of oncogenes and tumor suppressor genes, altered expression of microRNAs that regulate the expression of these genes occurs in many cancers. MicroRNA genes consist of a single RNA strand of approximately 21 to 23 nucleotides that does not encode proteins. They bind to messenger RNAs that contain complementary sequences and can block protein translation (32). MicroRNAs can function as either tumor suppressors or oncogenes and the disruption of normal microRNA function can occur through genetic changes (e.g., mutation, amplification), epigenetic silencing, or dysregulation by transcription factors. Approaches are being developed to target microRNAs involved in tumorigenesis.
Invasion and Metastasis
Metastasis is a process by which cancer cells spread from the primary tumor to distant sites (33). Cancer metastasis can proceed only if a series of sequential steps are completed, including proliferation, angiogenesis, invasion, embolisation or circulation, transportation, adherence in organs, adherence to vessel wall, and extravasation (Fig. 1.8).
Most types of cancer have an organ-specific pattern of metastasis. The propensity of various types of cancer to form metastases in specific organs was first proposed by Paget, who hypothesized that these patterns resulted from the “dependence of the seed (cancer cell) on the soil (the metastatic site)” (34). This hypothesis was suggested by the nonrandom pattern of metastasis. Paget concluded that metastases formed only when the seed and soil were compatible. It is now appreciated at a molecular level that metastasis is dependent on a balance between stimulating factors from both the tumor and host cells versus inhibitory signals. To produce metastasis, the balance must be weighted toward the stimulatory signals.
Cancer progression is a product of an evolving crosstalk between different cell types within the tumor and its surrounding supporting tissue, the tumor stroma (35). The tumor stroma contains a specific extracellular matrix as well as cellular components such as fibroblasts, immune and inflammatory cells, and blood vessel cells. The interactive signaling between tumor and stroma contributes to the formation of a complex multicellular organ. The organ microenvironment can markedly change the gene-expression patterns of cancer cells and therefore their behavior and growth potential (35). Recent studies regarding chemokines and their receptors provide important clues regarding why some cancers metastasize to specific organs. For example, breast cancer cells frequently express chemokine receptors CXCR4 and CCR7 at high levels. The specific ligands for these receptors, CXCL12 and CCL 21, are found at high levels in lymph nodes, lung, liver, and bone marrow, which are common sites for breast cancer metastasis.
Angiogenesis
All cells require oxygen and other nutrients for survival and growth, and cells must reside within 100 μm of a capillary in order to receive oxygen (36). Therefore, growth of new vessels, termed angiogenesis, is required for sustained malignant growth beyond approximately 1 mm in diameter (Fig. 1.9). Angiogenesis occurs as a result of a shift in balance toward proangiogenic factors within the tumor microenvironment along with down regulation of antiangiogenic influences. One of the primary mediators of angiogenesis is vascular endothelial growth factor A (VEGF-A) (37), which increases vascular permeability, stimulates endothelial cell proliferation and migration, and promotes endothelial cell survival (38). Other mediators of angiogenesis include tumor-derived factors and host stromal factors including interleukin-8, alpha v-beta 3 integrin, the tyrosine kinase receptor EphA2, and matrix metalloproteinases (39). From a translational perspective, patient-specific tumor microenvironmental characteristics may influence the response to antiangiogenic therapy (40). Therapeutic strategies to target angiogenesis include VEGF-A neutralizing antibodies (Bevacizumab) and multikinase inhibitors that target the VEGF-receptors along with other kinases. Bevacizumab is widely used in the treatment of metastatic colorectal cancer, advanced nonsmall cell lung cancer, glioblastoma, and metastatic renal cell carcinoma. Sorafenib and pazopanib are two examples of multikinase inhibitors that are approved for the treatment of metastatic renal cell carcinoma. There has been much enthusiasm for these drugs in the treatment of ovarian cancer; however, their benefit appears limited to improving time to progression with no overall survival benefit.
Invasion
Invasion through the basement membrane is a critical first step in metastasis and the primary feature that defines malignancy. Invasion requires the interplay between cancer cells and a permissive underlying stroma (41). Invasion of malignant cells through the basement membrane and endothelial cell migration for angiogenesis require degradation of the extracellular matrix. This process is facilitated by a group of enzymes called matrix metalloproteinases (MMPs), which are a family of zinc-dependent endopeptidases that digest collagen and other extracellular matrix components. They also stimulate proliferation and induce release of VEGF. Ovarian tumors overexpress MMP-2 and MMP-9, and this increased expression correlates with aggressive clinical features (42).
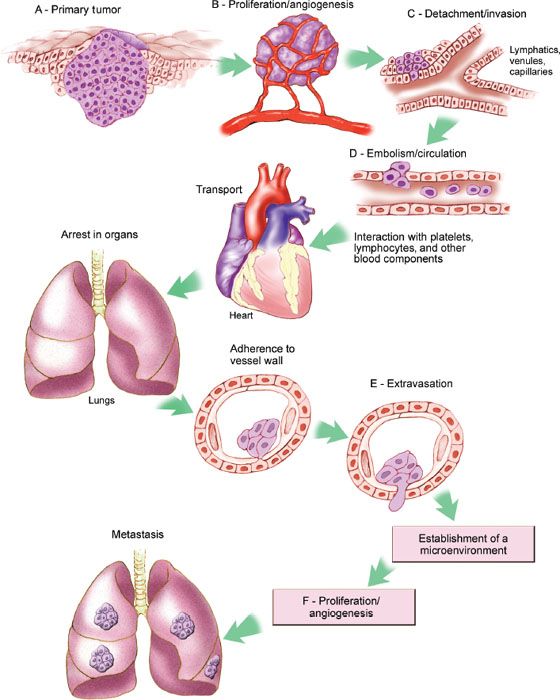
Figure 1.8 Molecular pathways involved in invasion and metastasis. (Redrawn from Fidler IJ. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat Rev Cancer. 2003;3:453–458.)
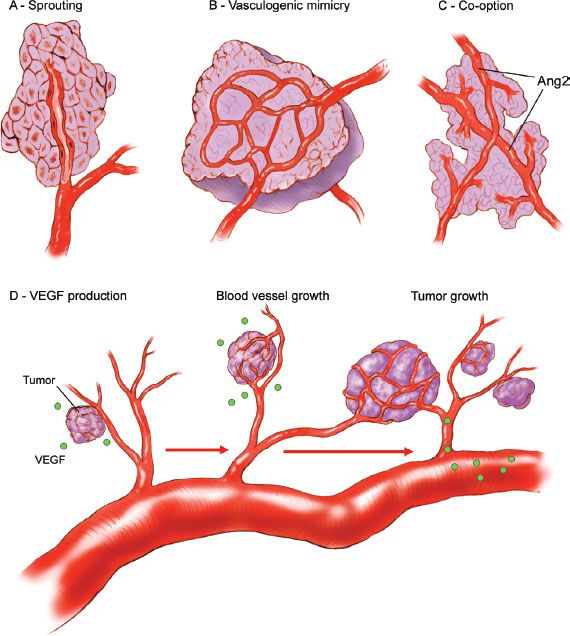
Figure 1.9 Mechanism of tumor neovascularization. A: Endothelial sprouting results from luminal endothelial cells migrating through the vessel basement membrane into underlying extracellular matrix, which is the dominant process for vessel growth. B: Aggressive tumor cells can form microvascular channels in a process termed “vasculogenic mimicry.” C: Tumors may co-opt pre-existing host tissue vasculature. D: A shift in the balance favoring the release of pro-angiogenic factors (e.g., VEGF) leads to endothelial activation, blood vessel growth, and tumor expansion. (Redrawn from Spannuth WA, Sood AK, Coleman RL. Angiogenesis as a strategic target for ovarian cancer therapy. Nat Clin Pract Oncol. 2008;5:194–204.)
Adhesion
Tumor cell adhesion to the extracellular matrix within tissues greatly influences the ability of a malignant cell to invade and metastasize (43). Given the shedding nature of ovarian cancer, adhesion molecules such as focal adhesion kinase, integrins, and E-cadherin have been evaluated for their role in peritoneal metastasis (44). The proteins of the extracellular matrix consist of type I and IV collagens, laminins, heparin sulfate proteoglycan, fibronectin, and other noncollagenous glycoproteins (45). Cell adhesion to these proteins is mediated in part by a group of heterodimeric transmembrane proteins called integrins, which are composed of a noncovalently associated α and β subunit that define the integrin–ligand specificity (46). Approximately 18 β subunits and eight α subunits have been identified, and at least 24 receptor combinations exist (47). The intracellular domains of integrins interact with cytoskeletal components and are actively involved in generating intracellular signals.
Cadherins are another group of cell–cell adhesion molecules that are involved in development and maintenance of solid tissues. E-cadherins are the subgroup predominantly found in epithelial cells (48). These transmembrane proteins mediate cell–cell adhesion: Cadherins on neighboring cells preferentially bind to the same types of cadherins on adjacent cells. E-cadherin is uniformly expressed in ovarian cancer, in low–malignant-potential tumors, in benign neoplasms, and—notably—in inclusion cysts of normal ovaries, but not in the normal surface epithelium (49). Cadherin dysfunction is associated with loss of cell–cell cohesion, altered cellular motility, and increased invasiveness and metastatic potential. Changes in the composition of the cadherin–catenin complex, phosphorylation of components in the complex and alterations in the interactions with the actin cytoskeleton have all been suggested as playing a role in regulating adhesion.
E-cadherin mutations occur only rarely (50), but cadherin expression may be downregulated in the absence of mutations. The cytoplasmic tails of cadherins exist as a macromolecular complex with β-catenin, which is involved in the WNT signaling pathways that regulate both adhesion and growth. Regulation of β-catenin activity also depends on the APC gene product and others in the WNT pathway. Mutations in the APC gene that abrogate its ability to inhibit β-catenin activity are common in the hereditary adenomatous polyposis coli syndrome and sporadic colon cancers (51). Likewise, mutations in the β-catenin gene that result in constitutively activated molecules have been observed in some cancers, including endometrial cancers (52).
Tumor Microenvironment
Within the tumor microenvironment, other cell types also play a critical role in tumor growth and progression. For example, certain types of inflammatory cells, including macrophages and mast cells and their associated cytokines, confer an unfavorable prognosis and increased tumor growth. Conversely, the presence of an adaptive immune response characterized by cytotoxic T cells is associated with improved clinical outcome. Cancer cells may evade immune recognition and destruction by various means, such as Fas ligand production to induce lymphocytic apoptosis and HLA-G secretion to inhibit natural-killer cell activity (53). Cytokine production by cancer cells promotes growth and inhibits apoptosis. However, the mechanistic relationships between the microenvironment and tumor growth remain only partially understood.
Immune checkpoints, crucial for maintaining immune self-tolerance and preventing auto-immune diseases, can be exploited by tumors to allow them to avoid immune surveillance (54). There are multiple costimulatory and inhibitory interactions that regulate T cell responses. Among them, cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) is expressed exclusively on T cells and downregulates T cell activation. CTLA4-blocking antibodies, such as ipilimumab, increase T-cell proliferation and activation, leading to improved anti-tumor responses. Ipilimumab is used in the treatment of malignant melanoma and is under evaluation for the treatment of multiple cancers.
Energy Metabolism
Cancer cells uptake increased amounts of glucose to satisfy their metabolic demands (55). Normal tissues generate energy using mitochondrial oxidative phosphorylation and switch to breaking down glucose only to derive energy in the absence of oxygen, which leads to the accumulation of lactate. In contrast, glycolysis of glucose to lactate occurs in cancers even in the presence of oxygen, a phenomenon called “aerobic glycolysis.” This is referred to as the Warburg effect, in honor of its discoverer (56). Because cancers often outgrow their blood supply and become hypoxic, the ability to survive using aerobic glycolysis instead of oxidative phosphorylation may be selected for during malignant transformation. Aerobic glycolysis may serve the increased metabolic requirement for carbon atoms to produce the macromolecules needed to build new cancer cells. If glucose is completely broken down to carbon dioxide via the citric acid cycle, these carbon building blocks are lost.
The hypoxia-inducible transcription factor-1α (HIF-1α) plays an important role in the cellular response to decreased oxygen in the local environment. When oxygen is absent, HIF-1α accumulates and promotes transcription of pro-angiogenesis genes as well as those involved in glucose transport and glycolysis. HIF-1α accumulation may be a consequence of loss of the VHL tumor suppressor gene, providing a link between the loss of a tumor suppressor and the altered metabolic phenotype of malignant cells. Another link between glucose metabolism and cancer is the finding that isocitrate dehydrogenase (IDH), an enzyme involved in glycolysis, is frequently mutated in glioblastomas. The common amino acid changing IDH mutations result in increased production of 2-hydroxyglutarate, which accumulates to high levels and plays a role in the development of these cancers.
Differences in metabolism between normal and malignant cells represent an appealing therapeutic target. In this regard, there is a suggestion that the diabetic drug metformin may have efficacy in the treatment and prevention of cancer. This appears to be independent of blood glucose level and the exact mechanism is unclear.
DNA Repair
Loss of DNA repair activity increases the likelihood of mutations being fixed in the genome and this is a hallmark of many cancers. Thousands of mutations occur in humans on a daily basis (57). Cells in many organs such as the skin, gastrointestinal tract, and respiratory tract that are exposed most directly to the environment constantly undergo renewal with shedding of differentiated cells that may contain mutations. Mammalian cells have highly evolved and complex DNA repair systems to maintain the integrity of the genome. A series of cell cycle checkpoints exist that allow the opportunity to pause for successful DNA repair, or alternatively for cell death if repair cannot be accomplished. DNA damage checkpoints occur at the boundaries between G1/S and G2/M and during S phase and mitotic spindle assembly. These checkpoints serve to protect against genetic damage that can lead to malignant transformation being fixed in the genome.
There are several repair mechanisms that operate on specific types of DNA damage during these checkpoints (55,56,58,59). Mismatch repair (MMR) excises nucleotides that are incorrectly paired with the correct nucleotide on the opposite DNA strand. Inherited mutations in MMR genes are responsible for Lynch syndrome. The nucleotide excision repair (NER) and base excision repair (BER) pathways respond to damage caused by DNA damaging agents. Mutations in the POLE gene have been found in a subset of endometrial cancers with the highest mutation rates. HR is a process that provides high-fidelity repair of complex DNA damage such as DNA cross-links, double-strand breaks, single-strand DNA gaps, and DNA interstrand cross-links. Inherited mutations in BRCA1–2 and other genes in the HR pathway cause familial breast/ovarian cancer susceptibility.
Gynecologic Malignancies
Gynecologic cancers vary with respect to grade, histology, stage, response to treatment, and survival. This clinical heterogeneity is attributable to differences in underlying molecular pathogenesis. Some cancers arise in a setting of inherited mutations in cancer susceptibility genes, but most occur sporadically in the absence of a strong hereditary predisposition. The spectrum of genes that are mutated varies between cancer types. For each type of cancer, there are a few genes that are frequently mutated, while a wider spectrum are altered in a small fraction of cases (13).
There is significant variety with respect to the spectrum of genetic changes within a given type of cancer. Cancers with a similar microscopic appearance may differ greatly at the molecular level. In some instances, molecular features may be predictive of clinical phenotypes such as stage, histologic type, and survival. With a more complete understanding of the clinical implications of various genetic alterations in gynecologic cancers, the molecular profile may prove valuable in predicting clinical behavior and response to treatment.
Endometrial Cancer
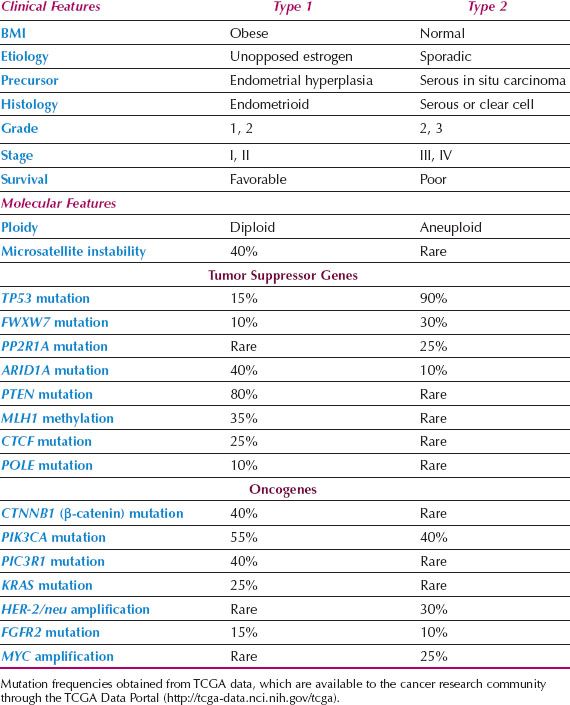
Epidemiologic and clinical studies of endometrial cancer have suggested that there are two distinct types of endometrial cancer (60). Type I cases are associated with unopposed estrogen stimulation and often develop in a background of endometrial hyperplasia. Obesity is the most common cause of unopposed estrogen and is part of a metabolic syndrome that includes insulin resistance and overexpression of insulin-like growth factors that may play a role in carcinogenesis. Type I cancers are well differentiated, endometrioid, early stage lesions, and have a favorable outcome. In contrast, type II cancers are poorly differentiated, often nonendometrioid (serous, clear cell), and are more virulent. Some may arise from serous carcinoma in situ of the endometrium. They often present at an advanced stage and survival is relatively poor. In practice, not all cancers can be neatly characterized as either pure type I or II lesions either clinically or based on their underlying genetic mutations (Table 1.3).
Similar to other human cancers, endometrial cancers are believed to arise because of a series of genetic alterations that result in progression from normal to precancer to invasive cancer. Unopposed estrogenic stimulation may contribute to the development of endometrial cancer through its mitogenic effect on the endometrium. A higher rate of proliferation in response to estrogens may lead to an increased frequency of spontaneous mutations. In addition, when genetic damage occurs, regardless of the cause, the presence of estrogens may facilitate clonal expansion. Estrogens may act as “complete carcinogens” that not only promote carcinogenesis by stimulating proliferation but also act as initiating agents by virtue of their carcinogenic metabolites. In contrast, progestins oppose the action of estrogens by downregulating estrogen receptor levels, decreasing proliferation, and increasing apoptosis. A small minority of endometrial cancers occur in women with a strong hereditary predisposition because of germline mutations in DNA repair genes causing Lynch syndrome.
Table 1.3 Characteristics of Type 1 versus Type 2 Endometrial Cancers
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


