specific helper T-cells. They become unable to respond to subsequent antigenic stimulation and may be excluded from lymphoid follicles.6
TABLE 29.1 CLASSIFICATION OF IMMUNE HEMOLYTIC ANEMIAS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
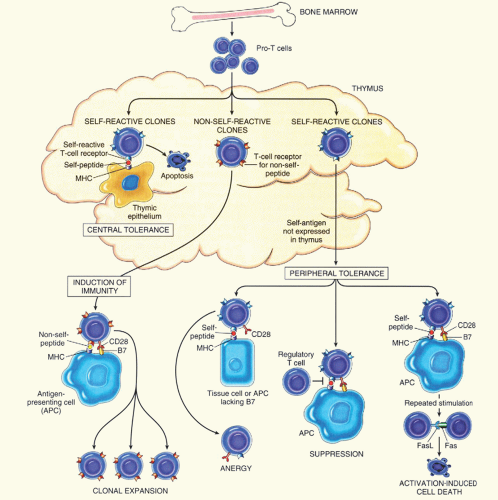 FIGURE 29.1. Schematic illustration of the mechanisms involved in central and peripheral tolerance. The principal mechanisms of tolerance in CD4+ T-cells are shown. APC: antigen-presenting cell. (Used with permission from Abbas AK, Diseases of immunity. In: Kumar V, Abbas A, Fausto N, eds. Robbins and Cotran pathologic basis of disease, 7th ed. Philadelphia, PA: Elsevier Saunders, 2005:223-225.) |
been implicated as environmental triggers of autoimmunity, possibly because of antigenic mimicry leading to tolerance breakdown, i.e., environmental or infectious agents may have molecular structures similar to self-antigens. Other possible mechanisms involve production of interferon γ during viral infection that causes up-regulation of FcRI. Alternatively, viral infection may cause a change in the expression pattern of Fc receptors as a result of transcriptional activation or other mechanisms (discussed further under section “IgG-Mediated Red Blood Cell Destruction”).9
TABLE 29.2 RED CELL DESTRUCTION BY IgM AND IgG ANTIBODIES | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
hemolysis (IgG3> IgG1> IgG4>>> IgG2).17, 18 The critical role of Fcγ receptors in immune destruction in vivo is further demonstrated by the therapeutic approach to management of AIHA and immune thrombocytopenic purpura (ITP). Treatment with corticosteroids, intravenous IgG (IVIG), anti-D, and/or splenectomy is aimed at reducing the capacity of reticuloendothelial cells to mediate immune clearance of IgG-sensitized RBCs.19 Administration of monoclonal antibody 2.4G2 in mice, which binds to and blocks mouse FcγRII and FcγRIII, allows rapid recovery after induction of AIHA, suggesting that alteration of the balance of stimulatory to inhibitory Fcγ receptors has a marked effect on disease progression and susceptibility. Understanding the detailed structure of the Fcγ receptors may lead to the development of novel therapeutic strategies. For example, molecules that inhibit the binding of the FcγR to an IgG-coated RBC or those that might inhibit the Fcγ receptor signaling at the different steps leading to phagocytosis may be useful therapeutic tools. Treatments that decrease expression of the activating Fcγ receptor or increase expression of the inhibitory Fcγ receptor may also be effective.9
TABLE 29.3 CHARACTERISTICS OF COMPLEMENT RECEPTORS | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
TABLE 29.4 CHARACTERISTICS OF FCγ RECEPTORS | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||
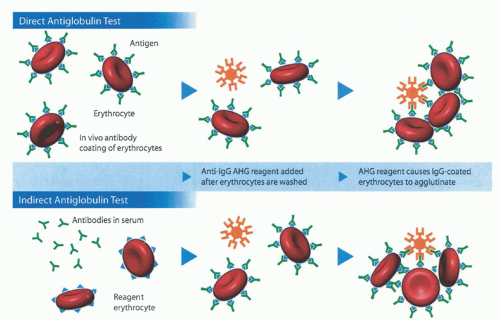 FIGURE 29.2. Direct antiglobulin test (DAT) and indirect antiglobulin test (IAT). The DAT reflects in vivo antibody sensitization of RBCs. Erythrocytes are washed to remove any unbound antibodies, and anti-IgG AHG reagent (AHG: antihuman globulin) is then added. IgG antibodies cannot cause direct RBC agglutination, but IgG-coated RBCs will agglutinate in the presence of AHG containing anti-IgG. This test can also be performed using anticomplement AHG reagent. If it is present, RBC-bound IgG can be eluted for specificity determination. The IAT is used to detect the presence of IgG antibodies in serum (in vitro sensitization). Reagent RBCs are incubated in the presence of serum that may contain antibodies. If they are present, antibodies bind to their target antigens on the reagent RBCs. After incubation, the RBCs are washed to remove unbound antibodies. Anti-IgG AHG reagent is added and will cause IgG-coated erythrocytes to agglutinate. (From Zarandona JM, Yazer MH. Teaching case report. The role of the Coombs test in evaluating hemolysis in adults. Can Med Assoc J 2006;174:305-307.) |
RBC-bound immunoglobulin and complement. The traditional tube method described above detects a lower limit of 150 to 200 IgG molecules per RBC.33 With PVP enhancement and an autoanalyzer, the detection limit decreases to as few as 8 IgG per RBC producing 5% agglutination. If bromelin is added as well, the sensitivity increases even further, with 1 IgG per RBC producing 5% agglutination and 3 IgG per RBC producing 50% agglutination.34 Additional acceptable techniques include flow cytometry, enzymelinked antiglobulin tests (ELISA), radioimmunoassays (RIA) using 125I-labeled anti-IgG or staphylococcal protein A, solid-phase techniques using microtiter plates, and gel testing.35 However, only the tube test, solid-phase test, and gel tests are in common use. Of these, the solid-phase and gel tests are the most standardized and have largely replaced older tube testing technology.
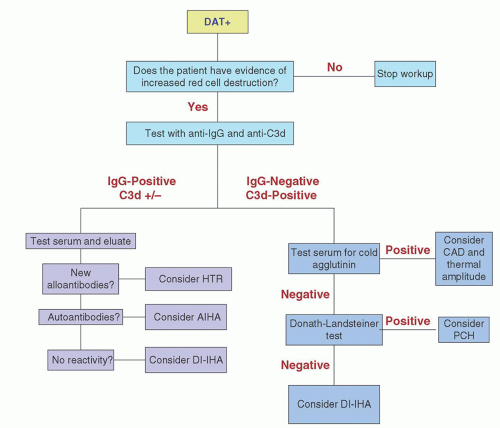 FIGURE 29.3. Flowchart suggesting a rational clinical laboratory approach for investigating a positive DAT. AIHA: autoimmune hemolytic anemia; CAD: cold agglutinin disease; DI-IHA: drug-induced immune hemolytic anemia; HTR: hemolytic transfusion reaction; PCH: paroxysmal cold hemoglobinuria. (Modified from Brecher ME, ed. Technical manual, 15th ed. Bethesda, MD: American Association of Blood Banks, 2005:480.) |
TABLE 29.5 COLD AUTOANTIBODIES | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 29.6 SEROLOGIC OVERVIEW OF HEMOLYTIC ANEMIAS | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
targets include Pr. Anti-Pr cold agglutinins tend to be high-titer, with a wide thermal range, and cause symptomatic anemia.55, 56 Other infrequent targets are Gd, Fl, Vo, Li, Sa, Lud, M, N, Me, Om, D, Sdx, and P.38, 44, 57 The fact that M. pneumoniae induces anti-I antibodies in the majority of patients is potentially related to the finding that sialylated I/i antigens serve as specific Mycoplasma receptors.58 Minor modification of this antigen may incite autoantibodies. Another theory suggests that an I-like antigen appears on the organism itself, and cross-reacting antibodies lead to RBC lysis.59 Despite the high rate of antibody production, clinically significant hemolysis occurs in very few patients.60 Infectious mononucleosis is also associated with CAD, but to a much lesser degree than Mycoplasma. Only 0.1% to 3.0% of mononucleosis patients have clinical hemolysis,61 although anti-i is present in 8% to 69% of sera post-infection.62, 63, 64 Therefore, the majority of patients with antibodies are asymptomatic. Anti-I activity is usually noted as well, but not to the same degree. Also, anti-Pr and anti-N have been reported.65 Both IgM and IgG antibodies as well as IgM rheumatoid-like factors reacting with IgG may act as cold agglutinins after infectious mononucleosis.66, 67 See Table 29.7 for a list of other infectious diseases associated with cold agglutinins, most of which are anti-I, although anti-i has been seen in cytomegalovirus infections and in lymphomas.68
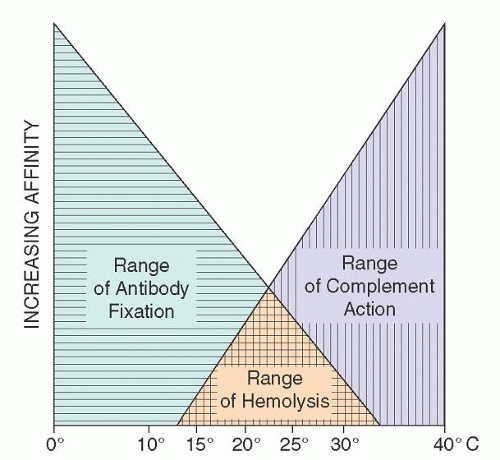 FIGURE 29.4. Temperature ranges for cold agglutinin fixation and lytic complement action. (From Schubothe H. The cold hemagglutinin disease. Semin Hematol 1966;3:27-47, with permission.) |
TABLE 29.7 SECONDARY COLD AGGLUTININ DISEASE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
obtained or unacceptable myelotoxicity results. Twice-weekly blood counts plus reticulocyte count should be monitored for toxicity.31, 75 Pulse therapy with cyclophosphamide (250 mg/day) and prednisone (100 mg/day ×4 days) every 2 to 3 weeks, blood counts permitting, or a large dose regimen of cyclophosphamide (1,000 mg), and intravenous methylprednisolone (500 mg) may adequately control hemolysis.76 In addition, α-interferon has reportedly produced significant remissions.77 However, recent studies have shown that rituximab is the most effective form of treatment and can be used as first line.
Related posts:
 Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Megaloblastic Anemias:Disorders of Impaired Dna Synthesis
Erythrocytosis
Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Megaloblastic Anemias:Disorders of Impaired Dna Synthesis
Erythrocytosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


