INTRODUCTION
SUMMARY
Acquired aplastic anemia is a clinical syndrome in which there is a deficiency of red cells, neutrophils, monocytes, and platelets in the blood, and fatty replacement of the marrow with a near absence of hematopoietic precursor cells. Reticulocytopenia is a constant feature. Neutropenia, monocytopenia, and thrombocytopenia, when severe, are life-threatening because of the risk of infection and bleeding, complicated by severe anemia. Most cases occur without an evident precipitating cause and are caused by autoreactive cytotoxic T lymphocytes that suppress or destroy primitive CD34+ multipotential hematopoietic cells. The disorder also can occur after (1) prolonged high-dose exposure to certain toxic chemicals (e.g., benzene), (2) after specific viral infections (e.g., Epstein-Barr virus), (3) as an idiosyncratic response to certain pharmaceuticals (e.g., ticlopidine, chloramphenicol), (4) as a feature of a connective tissue or autoimmune disorder (e.g., lupus erythematosus), or, (5) rarely, in association with pregnancy. The final common pathway may be through cytotoxic T-cell autoreactivity, whether idiopathic or associated with an inciting agent since they all respond in a similar fashion to immunosuppressive therapy. The differential diagnosis of acquired aplastic anemia includes a hypoplastic marrow that can accompany paroxysmal nocturnal hemoglobinuria or hypoplastic oligoblastic (myelodysplastic syndrome) or polyblastic myelogenous leukemia. Allogeneic hematopoietic stem cell transplantation is curative in approximately 80 percent of younger patients with high-resolution human leukocyte antigen–matched sibling donors, although the posttransplant period may be complicated by severe graft-versus-host disease. The disease may be significantly ameliorated or occasionally cured by immunotherapy, especially a regimen coupling antithymocyte globulin with cyclosporine. However, after successful treatment with immunosuppressive agents, the disease may relapse or evolve into a clonal myeloid disorder, such as paroxysmal nocturnal hemoglobinuria, a clonal cytopenia, or oligoblastic or polyblastic myelogenous leukemia. Several uncommon inherited disorders, including Fanconi anemia, Shwachman-Diamond syndrome, dyskeratosis congenita and others have as a primary manifestation aplastic hematopoiesis.
Acronyms and Abbreviations:
A, adenine; ALG, antilymphocyte globulin; ALL, acute lymphocytic leukemia; AML, acute myelogenous leukemia; ATG, antithymocyte globulin; ATR, ataxia-telangiectasia mutated and rad3-related kinase; BFU-E, erythroid burst-forming units; CD, cluster of differentiation; CFU-GM, colony-forming unit–granulocyte-macrophage; CMV, cytomegalovirus; EBV, Epstein-Barr virus; G, guanine; G-CSF, granulocyte colony-stimulating factor; HHV, human herpes virus; HLA, human leukocyte antigen; IL, interleukin; MRI, magnetic resonance imaging; PCP, pentachlorophenol; PNH, paroxysmal nocturnal hemoglobinuria; SCF, stem cell factor; T, thymine; TERC, telomerase RNA component; TERT, telomerase reverse transcriptase; TNF, tumor necrosis factor; TNT, trinitrotoluene; TPO, thrombopoietin.
ACQUIRED APLASTIC ANEMIA
Aplastic anemia is a clinical syndrome that results from a marked diminution of marrow blood cell production. The decrease in hematopoiesis results in reticulocytopenia, anemia, granulocytopenia, monocytopenia, and thrombocytopenia. The diagnosis usually requires the presence of pancytopenia with a neutrophil count fewer than 1500/μL (1.5 × 109/L), a platelet count fewer than 50,000/μL (50 × 109/L), a hemoglobin concentration less than 10 g/dL (100 g/L), and an absolute reticulocyte count fewer than 40,000/μL (40 × 109/L), accompanied by a hypocellular marrow without abnormal or malignant cells or fibrosis.1 For the purpose of therapeutic decision making, comparative clinical trials, and international sharing of data, the disease has been stratified into moderately severe, severe, and very severe acquired aplastic anemia based on the blood counts (especially the neutrophil count) and the degree of marrow hypocellularity (Table 35–1). Most cases of aplastic anemia are acquired; fewer cases are the result of an inherited disorder, such as Fanconi anemia, Shwachman-Diamond syndrome, and others (see “Hereditary Aplastic Anemia” below).
| Diagnostic Categories | Hemoglobin | Reticulocyte Concentration | Neutrophil Count | Platelet Count | Marrow Biopsy | Comments |
|---|---|---|---|---|---|---|
| Moderately severe | <100 g/L | <40 × 109/L | <1.5 × 109/L | <50 × 109/L | Marked decrease of hematopoietic cells. | At the time of diagnosis at least 2 of 3 blood counts should meet these criteria. |
| Severe | <90 g/L | <30.0 × 109/L | <0.5 × 109/L | <30.0 × 109/L | Marked decrease or absence of hematopoietic cells. | Search for a histocompatible sibling should be made if age permits. |
| Very severe | <80 g/L | <20.0 × 109/L | <0.2 × 109/L | <20.0 × 109/L | Marked decrease or absence of hematopoietic cells. | Search for a histocompatible sibling should be made if age permits. |
Aplastic anemia was first recognized by Paul Ehrlich in 1888.2 He described a young, pregnant woman who died of severe anemia and neutropenia. Thrombocytopenia was difficult to measure and the role of blood dust (platelets) was controversial at that time. Autopsy examination revealed a fatty marrow with essentially no hematopoiesis. The name aplastic anemia was subsequently applied to this disease by Chauffard, a French hematologist, in 1904,3 and although an anachronistic term because the morbidity is the result of pancytopenia, especially neutropenia and thrombocytopenia, the designation is entrenched in medical usage. For the next 40 years, many conditions that caused pancytopenia were confused with aplastic anemia based on incomplete or inadequate histologic study of the patient’s marrow.4 The development of improved instruments for percutaneous marrow biopsy in the last half of the 20th century improved diagnostic precision. In 1972, Thomas and his colleagues established that marrow transplantation from a histocompatible sibling donor could cure the disease.5 The disease initially was thought to result from an atrophy or chemical injury of primitive marrow hematopoietic cells. The unexpected recovery of marrow recipients who were given immunosuppressive conditioning therapy but who did not engraft with donor stem cells raised the possibility that the disease may not be intrinsic to primitive hematopoietic cells but the result of a suppression of hematopoietic cells by immune cells, notably T lymphocytes.6 The requirement to treat the recipient of a marrow transplant from an identical twin with immunosuppressive conditioning therapy for optimal results of transplant, buttressed this concept.7 This supposition was confirmed by a clinical trial that established antilymphocyte globulin (ALG) capable of ameliorating the disease in the majority of patients.8 Since that time, compelling evidence for a cellular autoimmune mechanism has accumulated (see “Etiology and Pathogenesis” below).
The International Aplastic Anemia and Agranulocytosis Study and a French study found the incidence of acquired aplastic anemia to be approximately 2 per 1,000,000 persons per year.1,9 This annual incidence has been confirmed in studies in Spain (Barcelona),10 Brazil (State of Parana),11 and Canada (British Columbia).12 The highest frequency of aplastic anemia occurs in persons between the ages of 15 and 25 years; a second peak occurs between the ages of 65 and 69.1 Aplastic anemia is more prevalent in the Far East where the incidence is approximately 7 per 1,000,000 in parts of China,13 approximately 4 per 1,000,000 in sections of Thailand,14 approximately 5 per 1,000,000 in areas of Malaysia,15 and approximately 7 per 1,000,000 among children of Asian descent living in Canada.12 The explanation for a twofold or greater incidence in the Orient compared to the Occident may be multifactorial,16 but a predisposition gene or genes is a likely component.12,17 Studies have not established the use of chloramphenicol in Asia as a cause. Poorly regulated exposure of workers to benzene is a factor,18 but the attributable risk from benzene and other toxic exposures does not explain the magnitude of the difference in the incidence in Asia compared to that in Europe and South America.16,17 A relationship to impure water use in Thailand has led to speculation of an infectious etiology, although no agent, including seronegative hepatitis, a known association with the onset of acquired aplastic anemia,16 has been identified. Seronegative viral hepatitis is a forerunner of approximately 7 percent of cases of acquired aplastic anemia.17,19 The male-to-female incidence ratio of aplastic anemia in most studies is approximately one.17
Table 35–2 lists the conditions associated with aplastic anemia.
| ACQUIRED |
| Autoimmune |
| Drugs |
| See Table 35–3 |
| Toxins |
| Benzene |
| Chlorinated hydrocarbons |
| Organophosphates |
| Viruses |
| Epstein-Barr virus |
| Non-A, -B, -C, -D, -E, or -G hepatitis virus |
| Human immunodeficiency virus (HIV) |
| Paroxysmal nocturnal hemoglobinuria |
| Autoimmune/connective tissue disorders |
| Eosinophilic fasciitis |
| Immune thyroid disease (Graves disease, Hashimoto thyroiditis) |
| Rheumatoid arthritis |
| Systemic lupus erythematosus |
| Thymoma |
| Pregnancy |
| Iatrogenic |
| Radiation |
| Cytotoxic drug therapy |
| INHERITED |
| Fanconi anemia |
| Dyskeratosis congenita |
| Shwachman-Diamond syndrome |
| Other rare syndromes (see Table 35–9) |
The final common pathway to the clinical disease is a decrease in blood cell formation in the marrow. The number of marrow CD34+ cells (multipotential hematopoietic progenitors) and their derivative colony-forming unit–granulocyte-macrophage (CFU-GM) and burst-forming unit–erythroid (BFU-E) are reduced markedly in patients with aplastic anemia.20,21,22,23 Long-term culture-initiating cells, an in vitro surrogate assay for hematopoietic stem cells, also are reduced to approximately 1 percent of normal values.23 Potential mechanisms responsible for acquired marrow cell failure include (1) cellular or humoral immune suppression of the marrow multipotential cells, (2) progressive erosion of chromosome telomeres, (3) direct toxicity to hematopoietic multipotential or stem cells, (4) a defect in the stromal microenvironment of the marrow required for hematopoietic cell development, and (5) impaired production or release of essential multilineage hematopoietic growth factors. There is little experimental evidence for a stromal microenvironmental defect or a deficit of critical hematopoietic growth factors or their receptors. Telomerase mutations with consequent telomere shortening may be involved in as many as 40 percent of patients.25 A susceptibility to the development of aplastic anemia is present in persons with certain human leukocyte antigen (HLA) types, such as HLA-DR15.25
Deficiencies in telomere repair could predispose to aplastic anemia by affecting the size of the multipotential hematopoietic cell compartment and by decreasing the multipotential cell’s response to marrow injury, and could play a role in the evolution of aplastic anemia to a clonal myeloid disease by contributing to genomic instability.24 Reduced hematopoiesis in most cases of aplastic anemia results from cytotoxic T-cell–mediated immune suppression of very early CD34+ hematopoietic multipotential progenitor or stem cells.26 A small fraction of cases is initiated by a toxic exposure, drug exposure, or viral infection, and in these cases the pathogenesis also may relate to autoimmunity as there is evidence of immune dysfunction in seronegative hepatitis, after benzene exposure, and many such patients respond to anti–T-cell therapy.26
In vitro and clinical observations have resulted in the identification of a cytotoxic T-cell–mediated attack on multipotential hematopoietic cells in the CD34+ cellular compartment as the basis for most cases of acquired aplastic anemia.27 Cellular immune injury to the marrow after drug-, viral-, or toxin-initiated marrow aplasia could result from the induction of neoantigens that provoke a secondary T-cell-mediated attack on hematopoietic cells. This mechanism could explain the response to immunosuppressive treatment in cases that follow exposure to an exogenous agent. Spontaneous or mitogen-induced increases in mononuclear cell production of interferon-γ,28,29 interleukin (IL)-2,29 and tumor necrosis factor-α (TNF-α)30,31 occur. These factors are inhibitory to hematopoietic cell development. Elevated serum levels of interferon-γ are present in 30 percent of patients with aplastic anemia, and interferon-γ expression has been detected in the marrow of most patients with acquired aplastic anemia.32 Addition of antibodies to interferon-γ enhances in vitro colony growth of marrow cells from affected patients.33 Long-term marrow cultures manipulated to elaborate exaggerated amounts of interferon-γ, markedly reduced the frequency of long-term culture-initiating cells.26 These observations indicate that acquired aplastic anemia is the result of cellular immune-induced apoptosis of primitive CD34+ multipotential hematopoietic progenitors, mediated by cytotoxic T lymphocytes, in part, through the expression of T-helper type 1 (Th1) inhibitory cytokines, interferon-γ, and TNF-α (Fig. 35–1).34 The secretion of interferon-γ is a result of the upregulation of the regulatory transcription factor T-bet,35 and apoptosis of CD34+ cells is, in part, mediated through a FAS-dependent pathway.26 Because HLA-DR2 is more prevalent in patients with aplastic anemia, antigen recognition may be a factor in those patients. A variety of other potential factors have been found in some patients, including nucleotide polymorphisms in cytokine genes, overexpression of perforin in marrow cells, and decreased expression of SLAM-associated protein (SAP), a modulator protein that inhibits interferon-γ secretion.26
Figure 35–1.
Immune pathogenesis of apoptosis of CD34 multipotential hematopoietic cells in acquired aplastic anemia. Antigens are presented to T lymphocytes by antigen-presenting cells (APCs). This triggers T cells to activate and proliferate. T-bet, a transcription factor, binds to the interferon-γ (IFN-γ) promoter region and induces gene expression. SLAM-associated protein (SAP) binds to Fyn and modulates the signaling lymphocyte activation molecule (SLAM) activity on IFN-γ expression, diminishing gene transcription. Patients with aplastic anemia show constitutive T-bet expression and low SAP levels. IFN-γ and tumor necrosis factor-α (TNF-α) upregulate both the T cell’s cellular receptors and the Fas receptor. Increased production of interleukin-2 leads to polyclonal expansion of T cells. Activation of the Fas receptor by the Fas ligand leads to apoptosis of target cells. Some effects of IFN-γ are mediated through interferon regulatory factor 1 (IRF-1), which inhibits the transcription of cellular genes and entry into the cell cycle. IFN-γ is a potent inducer of many cellular genes, including inducible nitric oxide synthase (NOS), and production of the toxic gas, nitric oxide (NO), may further diffuse the toxic effects. These events ultimately lead to reduced cell cycling and cell death by apoptosis. (Reproduced with permission from Young NS, Calado RT, Scheinberg P: Current concepts in the pathophysiology and treatment of aplastic anemia. Blood 2006 Oct 15;108(8):2509-2519.)
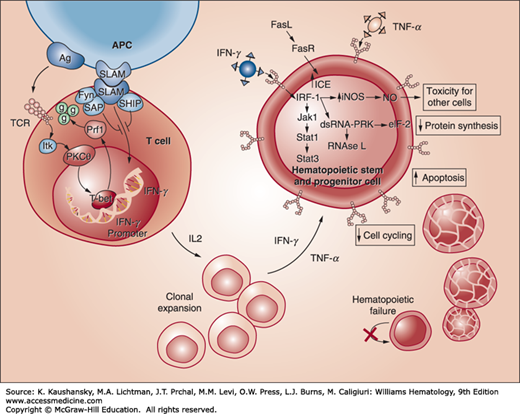
A decrease in regulatory T cells (CD4+CD25+FoxP3+) contributes to the expansion of an autoreactive CD8+CD28− T-cell population, which induces apoptosis of autologous hematopoietic multipotential hematopoietic cells.36,37,38 T-regulatory cells are a component of the immune system that suppress immune responses of other cells. They provide a “stop” for immune reactions that have achieved their purpose. They also play a role in preventing autoimmune reactions (Chap. 76). One mouse model of immune-related marrow failure, induced by infusion of parental lymph node cells into F1 hybrid recipients, caused a fatal aplastic anemia. The aplasia could be prevented by immunotherapy or with monoclonal antibodies to interferon-γ and TNF-α.26 Another mouse model of aplastic anemia induced by the infusion of lymph node cells histoincompatible for the minor H antigen, H60, resulted from the expansion of H60-specific CD8 T cells in recipient mice. The result was severe marrow aplasia. The effect of the CD8 T cells could be abrogated by either immunosuppressive agents or administration of CD4+CD25+ regulatory T cells,39 providing additional experimental evidence for the role of regulatory T cells in the prevention of aplastic anemia.
Several putative target antigens on affected hematopoietic cells have been identified. Autoantibodies to one putative antigen, kinectin, have been found in patients with aplastic anemia. T cells, responsive to kinectin-derived peptides, suppress granulocyte-monocyte colony growth in vitro. However, in these studies cytotoxic T lymphocytes with that specificity were not isolated from patients.40 Thus, the putative antigen(s) that is the target of the autoreactive T cells has not been identified.
A relationship between acquired aplastic anemia and hereditary aplastic anemia (Fanconi anemia or dyskeratosis congenita) in some patients has been suggested because the defects in telomerase and telomere repair, characteristic of Fanconi anemia and dyskeratosis congenita are shared in some adult patients with aplastic anemia, but in these cases there is no family history of such a disorder and no phenotypic abnormalities that characterize the hereditary disorders (see “Fanconi Anemia” and “Dyskeratosis Congenita” below). Telomeres shorten physiologically with age as telomerase becomes less active. T-cell–mediated acquired aplastic anemia is associated with telomere shortening which could reflect an inherited defect in telomerase or a senescent erosion of activity. The telomerase mechanism consists of a telomerase reverse transcriptase (TERT); an RNA template for TERT, the telomerase RNA component (TERC), and other stabilizing proteins.41,41a Cells with shortened telomeres normally undergo apoptosis unless DNA repair mechanisms are impaired allowing the development of aneuploidy and neoplastic transformation.
Chloramphenicol is the most notorious drug documented to cause aplastic anemia. Although this drug is directly myelosuppressive at very high dose because of its effect on mitochondrial DNA, the occurrence of aplastic anemia appears to be idiosyncratic, perhaps related to an inherited sensitivity to the nitroso-containing toxic intermediates.42 This sensitivity may produce immunologic marrow suppression, as a substantial proportion of affected patients respond to treatment with immunosuppressive therapy.43 The risk of developing aplastic anemia in patients treated with chloramphenicol is approximately 1 in 20,000, or 25 times that of the general population.44 Although its use as an antibiotic has been largely abandoned in industrialized countries, global reports of fatal aplastic anemia continue to appear with topical or systemic use of the drug.
Epidemiologic evidence established that quinacrine (Atabrine) increased the risk of aplastic anemia.45 This drug was administered to all U.S. troops in the South Pacific and Asiatic theaters of operations as prophylaxis for malaria during 1943 and 1944. The incidence of aplastic anemia was 7 to 28 cases per 1,000,000 personnel per year in the prophylaxis zones, whereas untreated soldiers had 1 to 2 cases per 1,000,000 personnel per year. The aplasia occurred during administration of the offending agent and was preceded by a characteristic rash in nearly half the cases. Many other drugs have been reported to increase the risk of aplastic anemia, but owing to incomplete reporting of information and the infrequency of the association, the spectrum of drug-induced aplastic anemia may not be fully appreciated. Table 35–3 is a partial list of drugs that have been implicated.46,47,48,49,50,51,52,53,54
| Category | High Risk | Intermediate Risk | Low Risk |
|---|---|---|---|
| Analgesic | Phenacetin, aspirin, salicylamide | ||
| Antiarrhythmic | Quinidine, tocainide | ||
| Antiarthritic | Gold salts | Colchicine | |
| Anticonvulsant | Carbamazepine, hydantoins, felbamate | Ethosuximide, phenacemide, primidone, trimethadione, sodium valproate | |
| Antihistamine | Chlorpheniramine, pyrilamine, tripelennamine | ||
| Antihypertensive | Captopril, methyldopa | ||
| Antiinflammatory | Penicillamine, phenylbutazone, oxyphenbutazone | Diclofenac, ibuprofen, indomethacin, naproxen, sulindac | |
| Antimicrobial | |||
| Antibacterial | Chloramphenicol | Dapsone, methicillin, penicillin, streptomycin, β-lactam antibiotics | |
| Antifungal | Amphotericin, flucytosine | ||
| Antiprotozoal | Quinacrine | Chloroquine, mepacrine, pyrimethamine | |
| Antineoplastic drugs | |||
| Alkylating agent | Busulfan, cyclophosphamide, melphalan, nitrogen mustard | ||
| Antimetabolite | Fluorouracil, mercaptopurine, methotrexate | ||
| Cytotoxic antibiotic | Daunorubicin, doxorubicin, mitoxantrone | ||
| Antiplatelet | Ticlopidine | ||
| Antithyroid | Carbimazole, methimazole, methylthiouracil, potassium perchlorate, propylthiouracil, sodium thiocyanate | ||
| Sedative and tranquilizer | Chlordiazepoxide, chlorpromazine (and other phenothiazines), lithium, meprobamate, methyprylon | ||
| Sulfa derivative | Sulfonamides | ||
| Antibacterial | Numerous sulfonamides | ||
| Diuretic | Acetazolamide | Chlorothiazide, furosemide | |
| Hypoglycemic | Chlorpropamide, tolbutamide | ||
| Miscellaneous | Allopurinol, interferon, pentoxifylline, penicillamine |
Many of these drugs are known to also induce selective cytopenias, such as agranulocytosis, which usually are reversible after discontinuation of the offending agent. These reversible reactions are not correlated with the risk of aplastic anemia, casting doubt on the effectiveness of routine monitoring of blood counts as a strategy to avoid aplastic anemia.
Because aplastic anemia is a rare event with drug use, it may occur because of an underlying metabolic or immunologic predisposition (gene polymorphism) in susceptible individuals. In the case of phenylbutazone-associated marrow aplasia, there is delayed oxidation and clearance of a related compound, acetanilide, as compared to either normal controls or those with aplastic anemia from other causes. This finding suggests excess accumulation of the drug as a potential mechanism for the aplasia. In some cases, drug interactions or synergy may be required to induce marrow aplasia. Cimetidine, a histamine H2-receptor antagonist, is occasionally implicated in the onset of cytopenias and aplastic anemia, perhaps owing to a direct effect on early hematopoietic progenitor cells.55 This drug accentuates the marrow-suppressive effects of the chemotherapy drug carmustine.56 In several instances, it has been reported as a possible cause of marrow aplasia when given with chloramphenicol.
There appears to be little difference in the age distribution, gender, response to immunotherapy, marrow transplantation, or survival, whether or not a drug exposure preceded the onset of the marrow aplasia.
Benzene was the first chemical linked to aplastic anemia, based on studies in factory workers before the 20th century.57,58,59 Benzene is used as a solvent and is employed in the manufacture of chemicals, drugs, dyes, and explosives. It has been a vital chemical in the manufacture of rubber and leather goods and has been used widely in the shoe industry, leading to an increased risk for aplastic anemia (and acute myelogenous leukemia) in workers exposed to a poorly regulated environment.59 In studies in China, aplastic anemia among workers was sixfold higher than in the general population.18
The U.S. Occupational Safety and Health Administration has lowered the permissible atmospheric exposure limit of benzene to 1 part per million (ppm) (8-hour time-weighted average) and short-term exposure to 5 ppm (15-minute time-weighted average). The National Institute for Occupational Safety and Health recommends limits of exposure of 0.1 ppm as the 8-hour weighted average and 1 ppm for 15-minute short-term exposure. Previous to that regulatory change, the frequency of aplastic anemia in workers exposed to greater than 100 ppm benzene was approximately 1 in 100 workers, which decreased to 1 in 1000 workers at 10 to 20 ppm exposure.58
Organochlorine and organophosphate pesticide compounds have been suspected in the onset of aplastic anemia60,61 and several studies have indicated an increased relative risk, especially for agricultural exposures11,16,62,63 and household11,63 exposures. These relationships are suspect because dose–disease relationships and other important factors have not been delineated, and several studies have not found an association with environmental exposures.12,64 DDT (dichlorodiphenyltrichloroethane), lindane, and chlordane are insecticides that also have been associated with cases of aplastic anemia.16,61 Occasional cases still occur following heavy exposure at industrial plants or after its use as a pesticide.65 Lindane is metabolized in part to pentachlorophenol (PCP), another potentially toxic chlorinated hydrocarbon that is manufactured for use as a wood preservative. Cases of aplastic anemia and related blood disorders have been attributed to PCP over the past 25 years.61,66 Prolonged exposures to petroleum distillates in the form of Stoddard solvent67 and acute exposure to toluene through the practice of glue sniffing68,69 also have been reported to cause marrow aplasia. Trinitrotoluene (TNT), an explosive used extensively during World Wars I and II, is absorbed readily by inhalation and through the skin.70 Fatal cases of aplastic anemia were observed in munitions workers exposed to TNT in Great Britain71 from 1940 to 1946. In most cases, these conclusions have not been derived from specific studies but from accumulation of case reports or from patient histories, making conclusions provisional, although the argument for minimizing exposures to potential toxins is logical in any case.
A relationship between hepatitis and the subsequent development of aplastic anemia has been the subject of a number of case reports, and this association was emphasized by two major reviews in the 1970s.72,73 In the aggregate, these reports summarized findings in more than 200 cases. In many instances, the hepatitis was improving or had resolved when the aplastic anemia was noted 4 to 12 weeks later. Approximately 10 percent of cases occurred more than 1 year after the initial diagnosis of hepatitis. Most patients were young (ages 18 to 20 years); two-thirds were male, and their survival was short (10 weeks). Although hepatitides A and B have been implicated in aplastic anemia in a small number of cases, most cases are related to non-A, non-B, non-C hepatitis.74,75,76 Severe aplastic anemia developed in 9 of 31 patients who underwent liver transplantation for non-A, non-B, non-C hepatitis, but in none of 1463 patients transplanted for other indications.77 Several lines of evidence indicate there is no causal association with hepatitis C virus, suggesting that an unknown viral agent is involved.16,78,79 Hepatitis virus B or C can be a secondary infection, if carefully screened blood products are not used for transfusion. In 15 patients with posthepatitic aplastic anemia, no evidence was found for hepatitis A, B, C, D, E, or G, transfusion-transmitted virus, or parvovirus B19.80 Several reports suggest a relationship of parvovirus B19 to aplastic anemia,81,82 whereas others have not.79 This relationship has not been established (Chap. 36). The effect of seronegative hepatitis may be mediated through an autoimmune T-cell effect because of evidence of T-cell activation and cytokine elaboration.26 These patients also have a similar response to combined immunotherapy as do those with idiopathic aplastic anemia83,84 (see “Treatment: Combination Immunotherapy” below).
Epstein-Barr virus (EBV) has been implicated in the pathogenesis of aplastic anemia.85,86 The onset usually occurs within 4 to 6 weeks of infection. In some cases, infectious mononucleosis is subclinical, with a finding of reactive lymphocytes in the blood film and serologic results consistent with a recent infection (Chap. 82). EBV has been detected in marrow cells,86 but it is uncertain whether marrow aplasia results from a direct effect or an immunologic response by the host. Patients have recovered following therapy with antithymocyte globulin.86
HIV infection frequently is associated with varying degrees of cytopenia. The marrow is often cellular, but occasional cases of aplastic anemia have been noted.87,88,89 Marrow hypoplasia may result from viral suppression and from the drugs used to control viral replication in this disorder. Human herpes virus (HHV)-6 has caused severe marrow aplasia subsequent to marrow transplantation for other disorders.90
The incidence of severe aplastic anemia was sevenfold greater than expected in patients with rheumatoid arthritis.52 It is uncertain whether the aplastic anemia is related directly to rheumatoid arthritis or to the various drugs used to treat the condition (gold salts, d-penicillamine, and nonsteroidal antiinflammatory agents). Occasional cases of aplastic anemia are seen in conjunction with systemic lupus erythematosus.91 In vitro studies found either the presence of an antibody92 or suppressor cell93,94 directed against hematopoietic progenitor cells. Patients have recovered after plasmapheresis,92 glucocorticoids,94 or cyclophosphamide therapy,93,95 which is compatible with an immune etiology.
Eosinophilic fasciitis, an uncommon connective tissue disorder with painful swelling and induration of the skin and subcutaneous tissue, has been associated with aplastic anemia.96,97 Although it may be antibody-mediated in some cases, it has been largely unresponsive to therapy.96 Nevertheless, (1) stem cell transplantation, (2) immunosuppressive therapy using cyclosporine, (3) immunosuppressive therapy using antithymocyte globulin (ATG), or (4) immunosuppressive therapy with ATG and cyclosporine has cured or significantly ameliorated the disease in a few patients.96,97
Severe aplastic anemia also has been reported coincident with immune thyroid disease (Graves disease)98,99,100,101,102 and the aplasia has been reversed with treatment of the hyperthyroidism. Aplastic anemia has occurred in association with thymoma.102,103,104,105,106,107,108 Autoimmune renal disease and aplastic anemia have occurred concurrently. The underlying relationship may be the role of cytotoxic T lymphocytes in the pathogenesis of several autoimmune diseases and in aplastic anemia.109
There are a number of reports of pregnancy-associated aplastic anemia, but the relationship between the two conditions is not always clear.110,111,112,113,114,115 In some patients, preexisting aplastic anemia is exacerbated with pregnancy, only to improve following termination of the pregnancy.110,111 In other cases, the aplasia develops during pregnancy with recurrences during subsequent pregnancies.111,112 Termination of pregnancy or delivery may improve the marrow function, but the disease may progress to a fatal outcome even after delivery.110,111,112 Therapy may include elective termination of early pregnancy, supportive care, immunosuppressive therapy, or marrow transplantation after delivery. Pregnancy in women previously treated with immunosuppression for aplastic anemia can result in the birth of a normal newborn.115 In this latter study of 36 pregnancies, 22 were uncomplicated, 7 were complicated by a relapse of the marrow aplasia, and 5 without marrow aplasia required red cell transfusion during delivery.115 One death occurred from cerebral thrombosis in a patient with paroxysmal nocturnal hemoglobinuria (PNH) and marrow aplasia.
Although marrow toxicity from cytotoxic chemotherapy or radiation produces direct damage to stem cells and more mature cells, resulting in marrow aplasia, most patients with acquired aplastic anemia cannot relate an exposure that would be responsible for marrow damage.
Chronic exposure to low doses of radiation or use of spinal radiation for ankylosing spondylitis is associated with an increased, but delayed, risk of developing aplastic anemia and acute leukemia.116,117 Patients who were given thorium dioxide (Thorotrast) as an intravenous contrast medium suffered numerous late complications, including malignant liver tumors, acute leukemia, and aplastic anemia.118 Chronic radium poisoning with osteitis of the jaw, osteogenic sarcoma, and aplastic anemia was seen in workers who painted watch dials with luminous paint when they moistened the brushes orally.119
Acute exposures to large doses of radiation are associated with the development of marrow aplasia and a gastrointestinal syndrome.120,121 Total-body exposure to between 1 and 2.5 Gy leads to gastrointestinal symptoms and depression of leukocyte counts, but most patients recover. A dose of 4.5 Gy leads to death in half the individuals (LD50) owing to marrow failure. Higher doses in the range of 10 Gy are universally fatal unless the patient receives extensive supportive care followed by marrow transplantation. Aplastic anemia associated with nuclear accidents was seen after the disaster that occurred at the Chernobyl nuclear power station in the Ukraine in 1986.122
Antineoplastic drugs such as alkylating agents, antimetabolites, and certain cytotoxic antibiotics have the potential for producing marrow aplasia. In general, this is transient, is an extension of their pharmacologic action, and resolves within several weeks of completing chemotherapy. Although unusual, severe marrow aplasia can follow use of the alkylating agent, busulfan, and may persist indefinitely. Patients may develop marrow aplasia 2 to 5 years after discontinuation of alkylating agent therapy. These cases often evolve into hypoplastic myelodysplastic syndromes.
Short-term clonal assays for marrow stromal cells have shown variable defects in stromal cell function in patients with aplastic anemia. Serum levels of stem cell factor (SCF) have been either moderately low or normal in several studies of aplastic anemia.123,124 Although SCF augments the growth of hematopoietic colonies from aplastic anemia patient’s marrows, its use in patients has not led to clinical remissions. Another early acting growth factor, FLT-3 ligand, is 30- to 100-fold elevated in the serum of patients with aplastic anemia, although the pathobiologic effect of this change is unclear.125 Fibroblasts grown from patients with severe aplastic anemia have subnormal cytokine production. However, serum levels of granulocyte colony-stimulating factor,126 erythropoietin,127 and thrombopoietin (TPO)128 are usually high. Synthesis of IL-1, an early stimulator of hematopoiesis, is decreased in mononuclear cells from patients with aplastic anemia.129 Studies of the microenvironment have shown relatively normal stromal cell proliferation and growth factor production.130 These findings, coupled with the limited response of patients with aplastic anemia to growth factors, suggest that cytokine deficiency is not the etiologic problem in most cases. The most compelling argument is that most patients transplanted for aplastic anemia are cured with allogeneic donor stem cells and autologous stroma.131
A rare exception to the negligible pathogenetic role of hematopoietic growth factors in the etiology of aplastic anemia is the homozygous or mixed heterozygous mutation of the TPO receptor gene, MPL, which can cause amegakaryocytic thrombocytopenia that evolves, later, into aplastic anemia (Chap. 117). Furthermore, eltrombopag, a TPO receptor agonist, can stimulate mono, or in some patients, bilineage or trilineage recovery of blood counts that may be sustained off therapy (see “Treatment: Cytokines” below).
The onset of symptoms of aplastic anemia may be gradual with pallor, weakness, dyspnea, and fatigue as a result of the anemia. Dependent petechiae, bruising, epistaxis, vaginal bleeding, and unexpected bleeding at other sites secondary to thrombocytopenia are frequent presenting signs of the underlying marrow disorder. Rarely, it may be more dramatic with fever, chills, and pharyngitis or other sites of infection resulting from severe neutropenia and monocytopenia. Physical examination generally is unrevealing except for evidence of anemia (e.g., conjunctival and cutaneous pallor, resting tachycardia) or cutaneous bleeding (e.g., ecchymoses and petechiae), gingival bleeding and intraoral purpura. Lymphadenopathy and splenomegaly are not features of aplastic anemia; such findings suggest an alternative diagnosis such as a clonal myeloid or lymphoid disease.
Patients with aplastic anemia have varying degrees of pancytopenia. Anemia is associated with a low reticulocyte index. The reticulocyte count is usually less than 1 percent and may be zero despite the high levels of erythropoietin. Absolute reticulocyte counts are usually fewer than 40,000/μL (40 × 109/L). Macrocytes may be present in the blood film and the mean cell volume (MCV) increased. The absolute neutrophil and monocyte count are low. An absolute neutrophil count fewer than 500/μL (0.5 × 109/L) along with a platelet count fewer than 30,000/μL (30 × 109/L) is indicative of severe disease, and a neutrophil count below 200/μL (0.2 × 109/L) denotes very severe disease (see Table 35–1). Lymphocyte production is thought to be normal, but patients may have mild lymphopenia. Platelets function normally. Significant qualitative changes of red cell, leukocyte, or platelet morphology on the blood film are not features of classical acquired aplastic anemia. On occasion, only one cell line is depressed initially, which may lead to an early diagnosis of pure red cell aplasia or amegakaryocytic thrombocytopenia. In such patients, other cell lines will fail shortly thereafter (days to weeks) and permit a definitive diagnosis. Table 35–4 is a plan for the initial laboratory investigation.
History and Physical Examination
|
The plasma contains high levels of hematopoietic growth factors, including erythropoietin, TPO, and myeloid colony-stimulating factors. Growth factor levels need not be measured, however, for clinical care. Plasma iron values are usually high, and 59Fe clearance is prolonged, with decreased incorporation into red cells.
The marrow aspirate typically contains numerous spicules with empty, fat-filled spaces, and relatively few hematopoietic cells. Lymphocytes, plasma cells, macrophages, and mast cells may be present. On occasion, occasional spicules are cellular or even hypercellular (“hot spots”), but megakaryocytes usually are reduced. These focal areas of residual hematopoiesis do not appear to be of prognostic significance. Residual granulocytic cells generally appear normal, but it is not unusual to see mild macronormoblastic erythropoiesis, presumably as a result of the high levels of erythropoietin. Marrow biopsy is essential to confirm the overall hypocellularity (Fig. 35–2), as a poor yield of spicules and cells occurs in marrow aspirates in other disorders, especially if fibrosis is present.
Figure 35–2.
Marrow biopsy in aplastic anemia. A. A normal marrow biopsy section of a young adult. B. The marrow biopsy section of a young adult with very severe aplastic anemia. The specimen is devoid of hematopoietic cells and contains only scattered lymphocytes and stromal cells. The hematopoietic space is replaced by reticular cells (pre-adipocytic fibroblasts) converted to adipocytes.
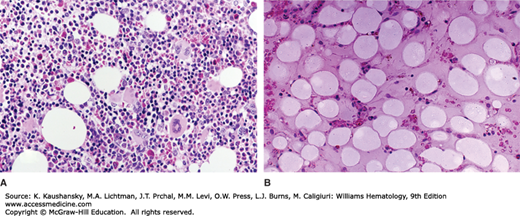
In severe aplastic anemia, as defined by the International Aplastic Anemia Study Group, less than 25 percent cellularity or less than 50 percent cellularity with less than 30 percent hematopoietic cells is seen in the marrow.
Cytogenetic analysis may be difficult to perform owing to low cellularity; thus, multiple aspirates may be required to provide sufficient cells for study. The results are normal in aplastic anemia. Clonal cytogenetic abnormalities in otherwise apparent aplastic anemia is indicative of an underlying hypoplastic clonal myeloid disease.132 The move to newer techniques such as microarray-based comparative genomic hybridization (CGH) permits detection of aneuploidies, deletions, duplications, and/or amplifications of any locus represented on an array. In addition, microarray-based CGH is an effective tool for the detection of submicroscopic chromosomal abnormalities. This approach would increase the sensitivity to detect chromosome abnormalities in very hypocellular marrow samples, compared to standard G-banding, despite dilution of scant hematopoietic cells with nonhematopoietic stromal cells (e.g., fibroblasts). Next-generation sequencing of targeted exons has uncovered 32 mutations associated with myeloid malignancies. These mutations occurred in nearly 20 percent (29 of 150 patients) of cases of aplastic anemia. These mutations include the genes ASXL1, DNMT3A, and BCOR, which are considered driver mutations in myelodysplastic syndrome and acute myelogenous leukemia. Seventeen of the 29 patients with one of these three mutations evolved to overt myelodysplasia.132a
Magnetic resonance imaging (MRI) can be used to distinguish between marrow fat and hematopoietic cells.133 This approach may be a more useful overall estimate of marrow hematopoietic cell density than morphologic techniques and may help differentiate hypoplastic myelogenous leukemia from aplastic anemia.128
Any disease that can present with pancytopenia may mimic aplastic anemia if only the blood counts are considered. Measurement of the reticulocyte count and an examination of the blood film and marrow biopsy are essential early steps to arrive at a diagnosis. A reticulocyte percentage of 0.5 percent to zero is strongly indicative of aplastic erythropoiesis, and when coupled with leukopenia and thrombocytopenia, points to aplastic anemia. Absence of qualitative abnormalities of cells on the blood film and a markedly hypocellular marrow are characteristic of acquired aplastic anemia. The disorders most commonly confused with severe aplastic anemia include the approximately 5 to 10 percent of patients with myelodysplastic syndromes who present with a hypoplastic rather than a hypercellular marrow. Myelodysplasia should be considered if there is abnormal blood film morphology consistent with myelodysplasia (e.g., poikilocytosis, basophilic stippling, neutrophils with hypogranulation or the pseudo–Pelger-Hüet anomaly). Marrow erythroid precursors in myelodysplasia may have dysmorphic features. Pathologic sideroblasts are inconsistent with aplastic anemia and a frequent feature of myelodysplasia. Granulocyte precursors may have reduced or abnormal granulation. Megakaryocytes may have abnormal nuclear lobulation (e.g., unilobular micromegakaryocytes; Chap. 87). If clonal cytogenetic abnormalities are found, a clonal myeloid disorder, especially myelodysplastic syndrome or hypocellular myelogenous leukemia is likely. MRI studies of bone may be useful in differentiating severe aplastic anemia from clonal myeloid syndromes. The former gives a fatty signal and the latter a diffuse cellular pattern.
A hypocellular marrow frequently is associated with PNH. PNH is characterized by an acquired mutation in the PIG-A gene that encodes an enzyme that is required to synthesize mannolipids. The gene mutation prevents the synthesis of the glycosylphosphatidylinositol anchor precursor. This moiety anchors several proteins, including inhibitors of the complement pathway to blood cell membranes, and its absence accounts for the complement-mediated hemolysis in PNH. As many as 50 percent of patients with otherwise typical aplastic anemia have evidence of glycosylphosphatidylinositol molecule defects and diminished phosphatidylinositol-anchored protein on leukocytes and red cells as judged by flow cytometry, analogous to that seen in PNH.134 The decrease or absence of these membrane proteins may make the PNH clone of cells resistant to the acquired immune attack on normal marrow components, or the phosphatidylinositol-anchored protein(s) on normal cells provides an epitope that initiates an aberrant T-cell attack, leaving the PNH clone relatively resistant (Chap. 40).26
Occasionally, apparent aplastic anemia may be the prodrome to childhood135 or, less commonly, adult136 acute lymphoblastic leukemia. Sometimes, careful examination of marrow cells by light microscopy or flow cytometry will uncover a population of leukemic lymphoblasts. In other cases, the acute leukemia may appear later. Hairy-cell leukemia, Hodgkin disease, or another lymphoma subtype, rarely, may be preceded by a period of marrow hypoplasia. Immunophenotyping of marrow and blood cells by flow cytometry for CD25 may uncover the presence of hairy cells. Other clinical features may be distinctive (Chap. 93). Organomegaly such as lymphadenopathy, hepatomegaly, or splenomegaly are inconsistent with the atrophic (hypoproliferative) features of aplastic anemia. Large granular lymphocytic leukemia has also been associated with aplastic anemia. Rare cases of typical acquired aplastic anemia have been followed by t(9;22)-positive acute lymphocytic leukemia (ALL) or chronic myelogenous leukemia (CML).136
RELATIONSHIP AMONG APLASTIC ANEMIA, PAROXYSMAL NOCTURNAL HEMOGLOBINURIA, AND CLONAL MYELOID DISEASES
In addition to the diagnostic difficulties occasionally presented by patients with hypoplastic myelodysplastic syndromes, hypoplastic acute myelogenous leukemia (AML), or PNH with hypocellular marrows, there may be a more fundamental relationship among these three diseases and aplastic anemia. The development of clonal cytogenetic abnormalities such as monosomy 7 or trisomy 8 in a patient with aplastic anemia portends the evolution of a myelodysplastic syndrome or acute leukemia. Occasionally, these cytogenetic markers have been transient, and in cases with disappearance of monosomy 7, hematologic improvement has occurred as well.137 Persistent monosomy 7 carries a poor prognosis as compared to trisomy 8.138,139
As many as 20 percent of patients with aplastic anemia have a 5-year probability of developing myelodysplasia.137 If one excludes any transformation to a clonal myeloid disorder that occurs up to 6 months after treatment to avoid misdiagnosis among the hypoplastic clonal myeloid diseases, the frequency of a clonal disorder was nearly 15 times greater in patients treated with immunosuppression as compared to those treated with marrow transplantation after 39 months of observation.140 This finding suggests either that immune suppression by anti–T-cell therapy enhances the evolution of a neoplastic clone or that it does not suppress the intrinsic tendency of aplastic anemia to evolve to a clonal disease, but provides the increased longevity of the patient required to express that potential. The latter interpretation is more likely as patients successfully treated solely with androgens develop clonal disease as frequently as those treated with immunosuppression.141 Transplantation may reduce the potential to clonal evolution in patients with aplastic anemia by reestablishing robust lymphohematopoiesis.
Telomere shortening also may play a pathogenetic role in the evolution of aplastic anemia into myelodysplasia. Patients with aplastic anemia have shorter telomere lengths than matched controls, and patients with aplastic anemia with persistent cytopenias had greater telomere shortening over time than matched controls. Three of five patients with telomere lengths less than 5 kb developed clonal cytogenetic changes, whereas patients with longer telomeres did not develop such diseases.23,142
The findings of mutated genes considered driver mutations in myelodysplastic syndrome or AML (see “Marrow Findings: Cytogenetic and Genetic Studies” earlier) in nearly 20 percent of a population of patients with clinical aplastic anemia indicates that clonal hematopoiesis may develop or be present surreptitiously. The precise relationships to the aplastic anemia lesion is uncertain but could be caused the outgrowth of a clone of cells in the background of severally suppressed polyclonal hematopoietic stem cells. These findings were more common in patients with a long duration of disease and with shorter telomeres.132a
The relationship of PNH to aplastic anemia remains enigmatic. Because hematopoietic stem cells lacking the phosphatidylinositol-anchored proteins are present in many or all normal persons in very small numbers,143 it is not surprising that more than 50 percent of patients with aplastic anemia may have a PNH cell population as detected by immunophenotyping.134 The probability of patients with aplastic anemia developing a clinical syndrome consistent with PNH is 10 to 20 percent, and this is not a consequence of immunosuppressive treatment.137 Patients also may present with the hemolytic anemia of PNH and later develop progressive marrow failure so that any pathogenetic explanation should consider both types of development of aplastic marrows in PNH. The PIG-A mutation may confer either a proliferative or survival advantage to PNH cells.144,145 A survival advantage could result if the anchor protein or one of its ligands served as an epitope for the T-lymphocyte cytotoxicity, which induces the marrow aplasia. In this case, the presenting event could either reflect cytopenias or the sensitivity of red cells to complement lysis and hemolysis, depending on the intrinsic proliferative potential of the PNH clone.
Within our current state of knowledge, aplastic anemia is an autoimmune process, and any residual hematopoiesis is presumably polyclonal. This is a critical distinction from hypoplastic leukemia and PNH, which are clonal (neoplastic) diseases. The environment of the aplastic marrow, however, may favor the eventual evolution of a mutant (malignant) clone, especially if immunotherapy is used, whereas hematopoietic stem cell transplantation may either ablate threatening minor clones or establish more robust hematopoiesis, an environment less conducive to clonal evolution.
Severe anemia, bleeding from thrombocytopenia, and, uncommonly at the time of diagnosis, infection secondary to granulocytopenia and monocytopenia require prompt attention to remove potential life-threatening conditions and improve patient comfort (Table 35–5). More specific treatment of the marrow aplasia involves two principal options: (1) syngeneic or allogeneic hematopoietic stem cell transplantation or (2) combination immunosuppressive therapy with ATG and cyclosporine. The selection of the specific mode of treatment depends on several factors, including the patient’s age and condition and the availability of a suitable allele-level HLA-matched hematopoietic stem cell donor. In general, transplantation is the preferred treatment for children and most otherwise healthy younger adults. Early histocompatibility testing of siblings is of particular importance because it establishes whether there is an optimal donor available to the patient for transplantation. The preferred stem cell source is a histocompatible sibling matched at the HLA-A, -B, -C, and -DR loci.
|
Although it has been recommended that red cell and platelet transfusions be used sparingly in potential transplant recipients to minimize sensitization to histocompatibility antigens, this has become less important since ATG and cyclophosphamide have been used as the preparative regimen for transplantation in aplastic anemia, as their use has markedly reduced the problem of graft rejection.146
Cytomegalovirus (CMV)-reduced risk red cells and platelets should be given to a potential transplant recipient to minimize problems with CMV infections after transplantation. Once a patient is shown to be CMV-positive, this restriction is no longer necessary. Leukocyte-depletion filters or CMV serotesting are equivalent methods of decreasing the risk of transmitting CMV.
Packed red cells to alleviate symptoms of anemia usually are indicated at hemoglobin values below 8 g/dL (80 g/L), unless comorbid medical conditions require a higher hemoglobin concentration. These products should be leukocyte-depleted to lessen leukocyte and platelet sensitization and to reduce subsequent transfusion reactions and radiated to reduce the potential for a transfusion-related graft-versus-host reaction. It is important not to transfuse patients with red cells (or platelets) from family members if transplantation within the family is remotely possible, as this approach may sensitize patients to minor histocompatibility antigens, increasing the risk of graft rejection after marrow transplantation. Following a marrow transplant, or in those individuals in whom transplantation is not a consideration, family members may be ideal donors for platelet products. Because each unit of red cells adds approximately 200 mg of iron to the total body iron, over the long-term transfusion-induced iron overload may occur. This is not a major problem in patients who respond to transplantation or immunosuppressive therapy, but it is an issue in nonresponders who require continued transfusion support. In the latter case, consideration should be given to iron-chelation therapy. Newer oral agents make this procedure easier to effect (Chap. 48).147
It is important to assess the risk of bleeding in each patient. Most patients tolerate platelet counts of 10,000/μL (10 × 109/L) without undue bruising or bleeding, unless a systemic infection is present or vascular integrity is impaired.148,149 A traumatic injury or surgery requires transfusion to greater than 50,000/μL or greater than 100,000/μL, respectively. Administration of ε-aminocaproic acid, 50 mg/kg per dose every 4 hours orally or intravenously, may reduce the bleeding tendency.150 Pooled random-donor platelets may be used until sensitization ensues, although it is preferable to use single-donor platelets from the onset to minimize sensitization to HLA or platelet antigens. Subsequently, single-donor apheresis products or HLA-matched platelets may be required.
Platelet refractoriness is a major problem with long-term transfusion support.151 This may occur transiently, with fever or infection, or as a chronic problem secondary to HLA sensitization. In the past, this occurred in approximately 50 percent of patients after 8 to 10 weeks of transfusion support. Filtration of blood and platelet concentrates to remove leukocytes reduces this problem to approximately 15 percent of patients receiving chronic transfusions.151,152 Patient’s should also get ABO-identical platelets because this enhances platelet survival and further decreases refractoriness to platelet transfusion. Single-donor HLA-matched apheresis-harvested platelets may be necessary in previously pregnant or transfused patients who are already allosensitized or who so become after treatment with leukoreduced platelets. The frequency of either of these events is less than 10 percent. Chapter 139 discusses approaches to chronic platelet transfusion.
Neutropenic precautions should be applied to hospitalized patients with a severe depression of the neutrophil count. The level of neutrophils requiring precautions is fewer than 500/μL (0.5 × 109/L). One approach is to use private rooms, with requirements for face masks and handwashing with antiseptic soap. Unwashed fresh fruits and vegetables should be avoided as they are sources of bacterial contamination. It is uncommon for patients with aplastic anemia to present with a significant infection. When patients with aplastic anemia become febrile, cultures should be obtained from the throat, sputum (if any), blood, urine, stool, and any suspicious lesions. Broad-spectrum bactericidal antibiotics should be initiated promptly, without awaiting culture results. The choice of antibiotics depends on the prevalence of organisms and their antibiotic sensitivity in the local setting. Organisms of concern usually include Staphylococcus aureus (notably methicillin-and oxacillin-resistant strains), Staphylococcus epidermidis (in patients with venous access devices), and Gram-negative organisms. Patients with persistent culture-negative fevers should be considered for antifungal treatment (Chap. 24).
In the past, leukocyte transfusions were used on a daily basis to reduce the short-term mortality from infections. It was unusual to detect more than 100 to 200 neutrophils per microliter for more than a few hours after transfusion. The yield of neutrophils can be increased by administering granulocyte colony-stimulating factor (G-CSF) to the donor,153
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


