Fred Y. Aoki Keywords acyclovir; amenamevir; cidofovir; cytomegalovirus; docosanol; Epstein-Barr virus; foscarnet; ganciclovir; helicase-primase inhibitors; herpes simplex virus; human herpesvirus 6; human herpesvirus 8; idoxuridine; maribavir; penciclovir; pritelivir; trifluorothymidine; valacyclovir; varicella-zoster virus
Antivirals against Herpes Viruses
The availability of efficacious and well-tolerated antiviral drugs that collectively inhibit the majority of the nine human herpesviruses (HHVs) has significantly reduced the morbidity and mortality caused by these viruses in healthy individuals. The availability of these drugs has also contributed to the control of herpesvirus infections resulting from immunosuppression caused by diseases, such as human immunodeficiency virus (HIV) infection, or reticuloendothelial system malignancies, such as Hodgkin’s disease. As important, the availability of these drugs has permitted the increasing and successful use of potent immunosuppressive agents for the management of a wide variety of diseases, such as transplant rejection, because a substantial proportion of herpesvirus infections are due to reactivation of asymptomatic latent herpesvirus infection. Those antiviral drugs, which are of established therapeutic effectiveness, as evidenced by registration by drug regulatory bodies in various countries, are listed in Table 45-1.
TABLE 45-1
Antiviral Agents of Established Therapeutic Effectiveness for Herpesvirus Infections
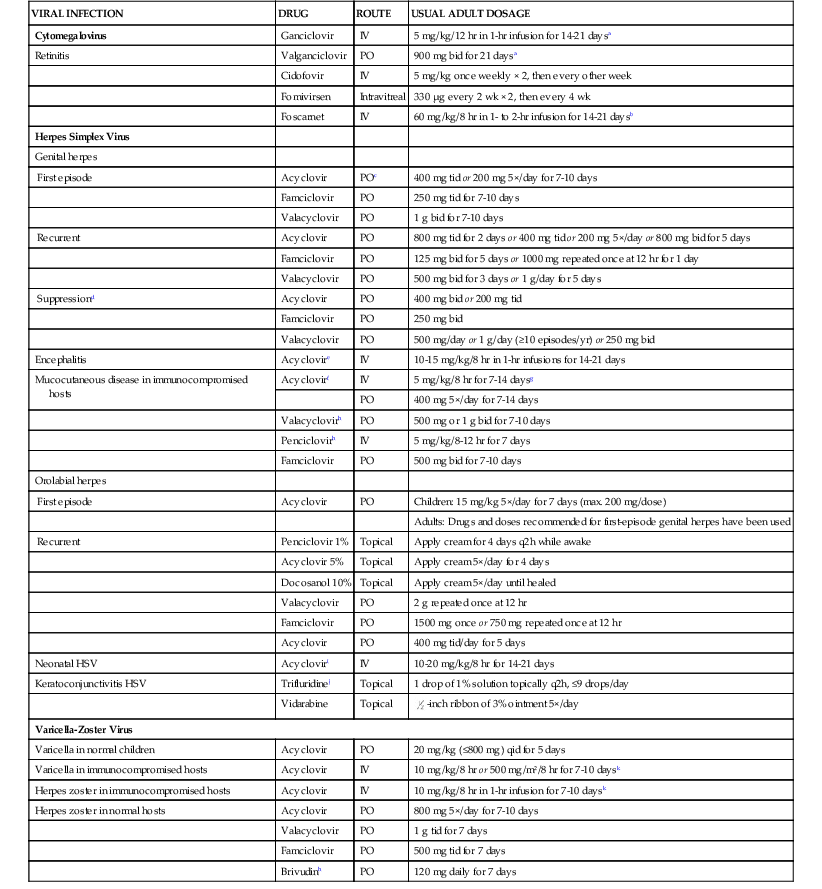
| VIRAL INFECTION | DRUG | ROUTE | USUAL ADULT DOSAGE |
| Cytomegalovirus | Ganciclovir | IV | 5 mg/kg/12 hr in 1-hr infusion for 14-21 daysa |
| Retinitis | Valganciclovir | PO | 900 mg bid for 21 daysa |
| Cidofovir | IV | 5 mg/kg once weekly × 2, then every other week | |
| Fomivirsen | Intravitreal | 330 µg every 2 wk × 2, then every 4 wk | |
| Foscarnet | IV | 60 mg/kg/8 hr in 1- to 2-hr infusion for 14-21 daysb | |
| Herpes Simplex Virus | |||
| Genital herpes | |||
| First episode | Acyclovir | POc | 400 mg tid or 200 mg 5×/day for 7-10 days |
| Famciclovir | PO | 250 mg tid for 7-10 days | |
| Valacyclovir | PO | 1 g bid for 7-10 days | |
| Recurrent | Acyclovir | PO | 800 mg tid for 2 days or 400 mg tid or 200 mg 5×/day or 800 mg bid for 5 days |
| Famciclovir | PO | 125 mg bid for 5 days or 1000 mg repeated once at 12 hr for 1 day | |
| Valacyclovir | PO | 500 mg bid for 3 days or 1 g/day for 5 days | |
| Suppressiond | Acyclovir | PO | 400 mg bid or 200 mg tid |
| Famciclovir | PO | 250 mg bid | |
| Valacyclovir | PO | 500 mg/day or 1 g/day (≥10 episodes/yr) or 250 mg bid | |
| Encephalitis | Acyclovire | IV | 10-15 mg/kg/8 hr in 1-hr infusions for 14-21 days |
| Mucocutaneous disease in immunocompromised hosts | Acyclovirf | IV | 5 mg/kg/8 hr for 7-14 daysg |
| PO | 400 mg 5×/day for 7-14 days | ||
| Valacyclovirh | PO | 500 mg or 1 g bid for 7-10 days | |
| Penciclovirh | IV | 5 mg/kg/8-12 hr for 7 days | |
| Famciclovir | PO | 500 mg bid for 7-10 days | |
| Orolabial herpes | |||
| First episode | Acyclovir | PO | Children: 15 mg/kg 5×/day for 7 days (max. 200 mg/dose) |
| Adults: Drugs and doses recommended for first-episode genital herpes have been used | |||
| Recurrent | Penciclovir 1% | Topical | Apply cream for 4 days q2h while awake |
| Acyclovir 5% | Topical | Apply cream 5×/day for 4 days | |
| Docosanol 10% | Topical | Apply cream 5×/day until healed | |
| Valacyclovir | PO | 2 g repeated once at 12 hr | |
| Famciclovir | PO | 1500 mg once or 750 mg repeated once at 12 hr | |
| Acyclovir | PO | 400 mg tid/day for 5 days | |
| Neonatal HSV | Acycloviri | IV | 10-20 mg/kg/8 hr for 14-21 days |
| Keratoconjunctivitis HSV | Trifluridinej | Topical | 1 drop of 1% solution topically q2h, ≤9 drops/day |
| Vidarabine | Topical |  -inch ribbon of 3% ointment 5×/day -inch ribbon of 3% ointment 5×/day | |
| Varicella-Zoster Virus | |||
| Varicella in normal children | Acyclovir | PO | 20 mg/kg (≤800 mg) qid for 5 days |
| Varicella in immunocompromised hosts | Acyclovir | IV | 10 mg/kg/8 hr or 500 mg/m2/8 hr for 7-10 daysk |
| Herpes zoster in immunocompromised hosts | Acyclovir | IV | 10 mg/kg/8 hr in 1-hr infusion for 7-10 daysk |
| Herpes zoster in normal hosts | Acyclovir | PO | 800 mg 5×/day for 7-10 days |
| Valacyclovir | PO | 1 g tid for 7 days | |
| Famciclovir | PO | 500 mg tid for 7 days | |
| Brivudinh | PO | 120 mg daily for 7 days | |
a In patients with acquired immunodeficiency syndrome (AIDS) or who are otherwise highly immunocompromised, long-term suppression with valganciclovir, 900 mg/day, is recommended after acute treatment. IV ganciclovir, 5 mg/kg given 7 day/wk or 6 mg/kg given 5 days/wk, or oral ganciclovir, 1 g tid are alternatives for suppression. These dosages are also approved for prevention of cytomegalovirus disease in transplant recipients.
b Long-term suppression with daily infusion of 90-120 mg/kg over 2 hr is recommended after initial treatment in patients with AIDS.
c In patients with severe initial genital herpes and in patients unable to tolerate oral medicines, IV acyclovir, 5 mg/kg/8 hr for 5-7 days, is recommended before a switch to an oral agent.
d Famciclovir, 500 mg bid, and valacyclovir, 500 mg bid, are effective in reducing recurrences in human immunodeficiency virus–infected patients.
e Higher dosages and 21 days of IV therapy are recommended by some authorities. The possible value of additional oral valacyclovir treatment afterward is under study.
f In acyclovir-resistant herpes simplex virus or varicella-zoster virus infections, IV foscarnet 40 mg/kg/8 hr seems beneficial. Duration of therapy depends on the clinical response. For limited cutaneous infections in immunocompromised patients, 5% acyclovir ointment can be applied to lesions every 3 hr, up to 6 times daily for 7 days (about  -inch ribbon per 4 inches2), using a finger cot or glove.
-inch ribbon per 4 inches2), using a finger cot or glove.
g Higher dosages (30 mg/kg/day) are recommended in progressive or visceral infections. Suggested pediatric dosage in children younger than 12 years is 10 mg/kg/8 hr for 7 days per manufacturer.
h Not approved by the U.S. Food and Drug Administration for this indication. IV penciclovir is unavailable in the United States.
i High dosage (20 mg/kg/8 hr) seems superior in disseminated and central nervous system disease in neonates.85
j An ophthalmic ointment of 3% acyclovir is available in some countries. Idoxuridine 0.1% solution, q1h while awake and q2h at night, or 0.5% ointment 5×/day is a less effective alternative. Treatment of herpes simplex virus ocular infections should be supervised by an ophthalmologist.
k Pediatric dosage of 500 mg/m2/8 hr for 7-10 days for children aged 1 year or older, although some experts recommend the 10-mg/kg/8 hr dosage for these children.111
HSV, herpes simplex virus; IV, intravenously; PO, orally.
Note: Please consult text and manufacturer’s product prescribing information for dosage adjustments in renal or hepatic insufficiency and in other circumstances.
Acyclovir and Valacyclovir
Spectrum
Acyclovir (9-[{2-hydroxyethoxy}methyl]-9H-guanine; acycloguanosine; Zovirax) is a deoxyguanosine analogue that has an acyclic side chain lacking the 3′-hydroxyl group instead of the cyclic ribose base of natural nucleosides (Fig. 45-1A). Valacyclovir (Valtrex) is the l-valyl ester prodrug of acyclovir. The clinically useful antiviral spectrum of acyclovir is limited to certain herpesviruses. Acyclovir is approximately 10 times more potent against herpes simplex virus type 1 (HSV-1) and herpes simplex virus type 2 (HSV-2) than against varicella-zoster virus (VZV), and it is even less active against cytomegalovirus (CMV) (Table 45-2).1,2 Acyclovir inhibits the replication of Epstein-Barr virus (EBV) in productively infected cells but does not affect latent or persistent infection. Acyclovir has shown antiviral activity in experimental HSV infection when administered topically, parenterally, or orally, and in simian varicella when given systemically.1 Enhanced antiherpesvirus activity occurs when acyclovir is given in combination with interferons (IFNs) and other antiviral agents in vitro and in animal models.1
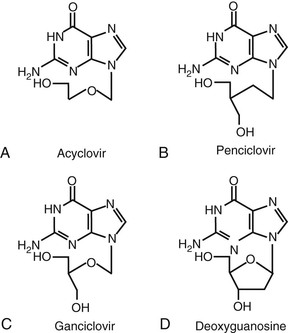
TABLE 45-2
Representative in Vitro Inhibitory Concentrations of Acyclic Nucleosides and Nucleotides for Clinical Isolates of Herpesviruses in Human Cells
| INHIBITORY CONCENTRATION (µg/mL) | ||||
| VIRUS | Acyclovir | Penciclovir | Ganciclovir | Cidofovir |
| Herpes simplex virus 1 | 0.02-1.9 | 0.2-1.8 | 0.05-0.6 | 0.4-3 |
| Herpes simplex virus 2 | 0.3-2.9 | 0.3-2.4 | 0.05-0.6 | 0.4-3 |
| Varicella-zoster virus | 0.8-5.2 | 0.9-5.1 | 0.2-2.8 | 0.25 |
| Cytomegalovirus | 2-57 | 52 | 0.2-2.8 | 0.2-0.9 |
| Epstein-Barr virus | 1.6 | — | 1.5 | <0.03 |
| Human herpesvirus type 6 | 7-23 | — | 0.3-7 | — |
| Human herpesvirus type 8 | 15-17 | — | 0.9-1.4 | 0.1-0.23 |
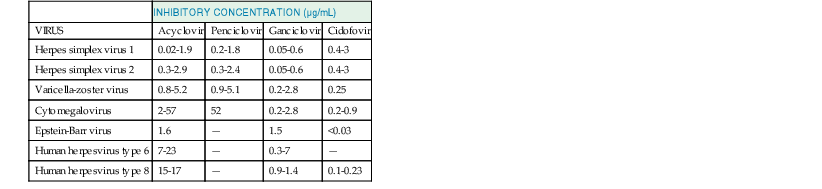
Data from references 6, 8, 14, 153, 156, 230, 232, 348, 393, 395, 494–496.
Growth of uninfected mammalian cells is generally unaffected by high acyclovir concentrations. Acyclovir (20 µg/mL) does not reproducibly alter cell-mediated immune responses of human peripheral blood leukocytes or affect human granulocyte progenitor cell growth in vitro.3
Mechanism of Action
Acyclovir is the prototype of a group of antiviral agents that are activated by viral thymidine kinase (TK) to become inhibitors of viral DNA polymerase that block viral DNA synthesis.1,2,4
Acyclovir uptake and intracellular phosphorylation to the monophosphate derivative are catalyzed by HSV TK. Cellular enzymes convert the monophosphate to acyclovir triphosphate, which is present in 40-fold to 100-fold higher concentrations in HSV-infected than in uninfected cells. Acyclovir triphosphate competitively inhibits viral DNA polymerase, and, to a much smaller extent, cellular DNA polymerases, with respect to deoxyguanosine triphosphate. Acyclovir triphosphate is also incorporated into viral DNA, where it acts as a chain terminator because of the lack of the 3′-hydroxyl group. Formation of a complex between the terminated DNA template containing acyclovir and the enzyme may lead to irreversible inactivation of the DNA polymerase. The DNA polymerases of various herpesviruses differ in their degree of inhibition by acyclovir triphosphate; the polymerases of EBV and CMV seem to be especially insensitive.
Resistance
Acyclovir-resistant HSV, often defined by an in vitro inhibitory concentration greater than 2 to 3 µg/mL, can be readily selected by passage in the presence of acyclovir and is present in native virus populations, with an approximate frequency of 1 in 103 to 104 infectious virions.4–6 Three basic resistance mechanisms have been identified: absent or low production of viral TK, altered TK substrate specificity (e.g., phosphorylation of thymidine, but not of acyclovir), and altered viral DNA polymerase. Changes in these viral enzymes relate to point mutations or base insertions or deletions in the corresponding genes.7,8 The most common mechanism found in clinical HSV isolates is absent or deficient TK activity.4,9 Less commonly, resistant isolates have altered TK activity, and DNA polymerase mutants are rare in clinical strains. Heterogeneous mixtures are commonly found. TK-negative variants are cross resistant to other agents activated by viral TK (e.g., penciclovir, ganciclovir), but TK-altered and DNA polymerase mutants variably retain susceptibility.4
The prevalence of acyclovir-resistant HSV isolates in immunocompetent hosts is about 0.1% to 0.7%, but increases to approximately 4% to 14% in immunocompromised patients.4,7,10 During several decades of use, no increase in the prevalence of acyclovir-resistant variants has occurred in immunocompetent individuals.4,11 Resistant HSV-2 has been found in 0.2% of HIV-negative and 5.3% of HIV-positive individuals with genital herpes,12 and it has been recovered from 11% to 17% of individuals with acquired immunodeficiency syndrome (AIDS) or recipients of allograft transplants receiving acyclovir treatment for 2 weeks or longer.13 Progressive HSV disease associated with recovery of acyclovir-resistant virus and poor response to acyclovir therapy is well recognized in immunocompromised patients. Painful ulcerating perirectal lesions, often indolent and necrotizing, caused by HSV-2 represent the most common pattern in patients with AIDS. Orofacial disease caused by HSV-1 is common in transplant recipients. The risk factors for resistance emergence include degree of immunosuppression, size of lesions, repeated or prolonged use of acyclovir for treatment rather than prophylaxis, and, possibly, the use of topical acyclovir in genital herpes.11
TK-negative HSVs are less neurovirulent than wild-type strains and are unable to reactivate from latency in animal models, although they may cause extensive mucocutaneous disease in immunocompromised hosts.4,6 TK-deficient, TK-altered, or DNA polymerase mutants have variable decreases in pathogenicity. Acyclovir-resistant HSV recurrent genital or ocular infections have rarely been found in immunocompetent hosts.4 One case of possible person-to-person spread of resistant HSV has been reported. Recurrences after cessation of acyclovir are usually caused by sensitive virus.4 In patients with AIDS, persistent shedding of resistant HSV at the site of initial infection and recurrences with acyclovir-resistant variants have been found in the absence of selective drug pressure.14 Visceral disease is uncommon, but pneumonitis, meningoencephalitis, esophagitis, hepatitis, retinal necrosis, and disseminated infection have occurred with resistant variants, including instances in neonates.4,12,15
Depending on the degree of immunosuppression, resistant HSV infections may undergo spontaneous healing during or after cessation of acyclovir therapy. In patients with progressive disease, intravenous foscarnet therapy is effective, but therapy with vidarabine is not.14 Intravenous cidofovir also seems to be effective. High-dose continuous infusion of acyclovir,16 topical trifluridine, topical IFN-α2 alone or in combination with topical trifluridine,17 topical foscarnet,18 topical cidofovir gel, and topical imiquimod19 have been used with variable success.20
Acyclovir resistance in VZV isolates, associated with 20-fold to 40-fold increases in inhibitory concentrations, is usually related to mutations in VZV TK with inability to phosphorylate acyclovir, or, less often, to mutations in viral DNA polymerase. Although rare, resistant isolates, including the Oka VZV vaccine strain,21 have been recovered from highly immunocompromised children and adults with chronic disseminated, hyperkeratotic, or verrucous papular lesions that failed to heal with intravenous acyclovir.22,23 Invasive disease with resistant variants occurs. Long-term suppressive therapy with subtherapeutic dosages of acyclovir seems to be a risk factor. Intravenous foscarnet or cidofovir may be effective for acyclovir-resistant VZV infections.24,25
Pharmacokinetics
The bioavailability of oral acyclovir is low (15% to 21%) and decreases with increasing dosages.1 Peak plasma concentrations average 0.4 to 0.8 µg/mL after 200-mg oral doses and increase to about 1.6 µg/mL with 800-mg doses. Bioavailability is lower in transplant recipients, in whom doses of 400 mg provide peak levels of 0.7 to 0.9 µg/mL. A liquid suspension has lower oral bioavailability; peak plasma concentrations average 1 µg/mL in children receiving dosages of 600 mg/m2. In neonates and infants younger than 2 years, oral bioavailability averages 12%, acyclovir kinetics are affected by prematurity and age younger than 1 month, and weight-adjusted dosing is essential.26 Peak and trough plasma concentrations average 9.8 µg/mL and 0.7 µg/mL, respectively, after intravenous administration of 5 mg/kg every 8 hours, and 20.7 µg/mL and 2.3 µg/mL, respectively, after 10 mg/kg every 8 hours. Peak concentrations average 10.3 µg/mL and 20.7 µg/mL after intravenous doses of 250 mg/m2 and 500 mg/m2, respectively, in children.
After oral administration, valacyclovir is readily absorbed, most likely via human peptide transporter 1 (hPEPT1), and rapidly converted to acyclovir during first-pass enzymatic hydrolysis in the liver and intestine by an enzyme designated valacyclovir hydrolase.27,28 The relative bioavailability of acyclovir is three to five times greater after ingestion of valacyclovir, whose absolute bioavailability averages 54% to 70%.29 Estimated bioavailability is 48% in hospitalized immunocompromised children 5 years old or older.30 Peak plasma levels of valacyclovir are 0.4 µg/mL or less after 1000-mg doses. Peak plasma acyclovir levels average 5 µg/mL and 8.5 µg/mL after doses of 1000 mg and 2000 mg in adults and are estimated to be 7 µg/mL to 8 µg/mL after doses of 30 mg/kg valacyclovir in children.29,31 Total acyclovir exposure with administration of valacyclovir is similar to that seen with intravenous acyclovir, although peak plasma concentrations are twofold to fourfold lower.30 In older adults, peak plasma concentrations increase 15% to 20%, and overall acyclovir exposure increases 30% to 50%,27 probably because of reduced renal clearance in this population.
Acyclovir is distributed widely in body fluids. Plasma protein binding is less than 20%. Concentrations over time in noninflamed cerebrospinal fluid (CSF) average 20% of those in serum.32 Salivary concentrations average 13% of plasma levels, but concentrations in vaginal secretions range from 15% to 170% of those in plasma. Zoster vesicular fluid levels are similar to those in plasma. Aqueous humor levels average 37% of concurrent plasma values. Acyclovir is concentrated in breast milk at approximately threefold higher levels than in maternal serum. Plasma levels in newborns are similar to maternal ones, and amniotic fluid and placental concentrations are severalfold higher.33 Percutaneous absorption of acyclovir after topical administration seems to be low.
The mean plasma elimination half-life ( ) of acyclovir is about 2.5 to 3 hours (range, 1.5 to 6.3 hours) in adults with normal renal function; it is slightly longer (3.8 hours) in neonates and increases to 19.5 hours in anuric patients.33,34 Renal excretion of unmetabolized acyclovir by glomerular filtration and tubular secretion accounts for 60% to 91% of an administered dose, whereas less than 15% is excreted as 9-carboxymethoxymethylguanine or minor metabolites.35 Acyclovir and valacyclovir dosage reductions are indicated in patients with a creatinine clearance (CrCl) of less than 50 mL/min (Tables 45-3 and 45-4). Hemodialysis removes 33% to 60% of acyclovir during a 6-hour session, whereas peritoneal dialysis removes very little.36 Dosing is recommended after hemodialysis, but supplementation is not needed during continuous ambulatory peritoneal dialysis. Bioavailability is about 61% after intraperitoneal dosing.37 Table 45-4 lists dosage adjustments for valacyclovir in patients with impaired renal function.
) of acyclovir is about 2.5 to 3 hours (range, 1.5 to 6.3 hours) in adults with normal renal function; it is slightly longer (3.8 hours) in neonates and increases to 19.5 hours in anuric patients.33,34 Renal excretion of unmetabolized acyclovir by glomerular filtration and tubular secretion accounts for 60% to 91% of an administered dose, whereas less than 15% is excreted as 9-carboxymethoxymethylguanine or minor metabolites.35 Acyclovir and valacyclovir dosage reductions are indicated in patients with a creatinine clearance (CrCl) of less than 50 mL/min (Tables 45-3 and 45-4). Hemodialysis removes 33% to 60% of acyclovir during a 6-hour session, whereas peritoneal dialysis removes very little.36 Dosing is recommended after hemodialysis, but supplementation is not needed during continuous ambulatory peritoneal dialysis. Bioavailability is about 61% after intraperitoneal dosing.37 Table 45-4 lists dosage adjustments for valacyclovir in patients with impaired renal function.
TABLE 45-3
Acyclovir Dosage Adjustments Suggested for Patients with Impaired Renal Function
| CREATININE CLEARANCE (mL/min/1.73 m2) | INTRAVENOUS | ORAL* | ||
| Standard Dose (%) | Dosing Interval (hr) | Dose (mg) | Dosing Interval (hr) | |
| >50 | 100 | 8 | 800 | 4 |
| 25-50 | 100 | 12 | 800 | 4 |
| 10-25 | 100 | 24 | 800 | 8 |
| 0-10† | 50 | 24 | 800 | 12‡ |
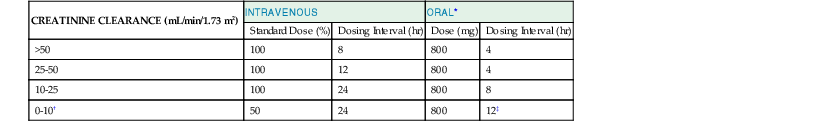
* Oral acyclovir dosage adjustments are needed for severe renal insufficiency. Recommendations are based on high-dose oral regimen (4000 mg/day). For the low-dose (1000 mg/day) oral regimen, the suggested dosage is 200 mg q12h when creatinine clearance is less than 10 mL/min/1.73 m2.
† An alternative in patients with end-stage renal disease is administration of 14% of standard dosage q8h after loading with 37% of the standard dosage. In hemodialysis, use 60% to 100% of standard dosage after the hemodialysis run only.
‡ In dialysis-dependent patients, a further dosage reduction to 200 mg/12 hr and 400 mg after dialysis is recommended to avoid toxic levels. A dosage of 800 mg PO q24h has been suggested for patients on continuous ambulatory peritoneal dialysis.36 For adults during continuous renal replacement therapy, give q48h.
Modified from Blum MR, Liao SH, de Miranda P. Overview of acyclovir pharmacokinetic disposition in adults and children. Am J Med. 1982;73:186-192; and Laskin OL, Longstreth JA, Whelton A, et al. Effect of renal failure on the pharmacokinetics of acyclovir. Am J Med. 1982;73:197-201.

Interactions
Severe somnolence and lethargy may occur with combinations of zidovudine and acyclovir.38 Concomitant use of cyclosporine, and probably of other nephrotoxic agents, enhances the risk for nephrotoxicity. Probenecid and cimetidine slow valacyclovir metabolism, decrease renal acyclovir clearance, and increase overall acyclovir exposure by 48% and 27%.39 By competing for the organic acid secretory pathway, acyclovir may decrease the renal clearance of other drugs eliminated by active renal secretion, such as methotrexate. Thiazide diuretics or the hPEPT1 substrate cephalexin do not substantially alter valacyclovir pharmacokinetics.27 No pharmacokinetic interaction was observed between tipranavir/ritonavir and single-dose valacyclovir at steady state in healthy volunteers.28
Toxicity
Topical acyclovir may cause transient burning when it is applied to genital lesions. The polyethylene glycol base of topical acyclovir may cause mucosal irritation and is not approved for intravaginal use. Acyclovir cream uncommonly causes allergic contact dermatitis.1
Intravenous acyclovir is generally well tolerated,1 although inflammation, phlebitis, and, rarely, vesicular eruption can occur at the injection site after extravasation of the alkaline solution (pH 9 to 11). Uncommon side effects include rash, diaphoresis, hematuria, hypotension, headache, and nausea. Approximately 1% to 4% of patients receiving intravenous acyclovir have manifested neurotoxicity, characterized by lethargy, confusion, obtundation, tremor, myoclonus, hallucinations, delirium, seizures, extrapyramidal signs, autonomic instability, or coma.40 Diffuse electroencephalographic abnormalities and increased CSF concentrations of myelin basic protein may occur. Symptoms of neurotoxicity usually develop within 1 to 3 days after starting treatment. Most of these patients have acute renal dysfunction or preexisting renal disease, and neurotoxicity occurs in association with high serum acyclovir concentrations (>25 µg/mL) and detectable CSF levels of 9-carboxymethoxymethylguanine, the main metabolite of acyclovir, which may be the cause of acyclovir neurotoxicity. It has been detected at higher concentrations in the CSF of subjects with neurotoxicity41 than in asymptomatic normal individuals or those with chronic kidney disease.31 Neurotoxicity occurs more often after valacyclovir. Neurologic side effects usually resolve within several days after drug concentrations decrease. Hemodialysis may be useful in severe cases.
Reversible renal dysfunction has been observed in approximately 5% of patients, and a higher proportion of children, treated with intravenous acyclovir.1 Acyclovir can cause a crystalline nephropathy and, rarely, interstitial nephritis.42 Acyclovir solubility decreases to 2.5 mg/mL at 37° C, and crystalluria has been described in adult and pediatric patients. Obstructive nephropathy may manifest as nausea, emesis, flank pain, and increasing azotemia. Co-administration with other nephrotoxic drugs, bolus infusion, dehydration, preexisting renal insufficiency, high doses, and high acyclovir plasma levels are risk factors. Nephrotoxicity usually resolves with drug cessation and volume expansion.
Oral acyclovir has been associated infrequently with nausea, diarrhea, rash, and headache, and uncommonly with renal insufficiency or neurotoxicity. Immediate hypersensitivity reactions to acyclovir are rare, but may be managed with oral desensitization.43 Long-term acyclovir suppression for frequently recurring genital or mucocutaneous infections seems well tolerated after long-term use,44,45 and no adverse effects on sperm production or peripheral blood lymphocyte cytogenetics have been detected.46 Oral acyclovir can cause neutropenia in infants, however.47 High-dose valacyclovir (8 g/day) is associated with gastrointestinal intolerance, azotemia, possibly thrombotic microangiopathy in patients with AIDS,48 and with confusion and hallucinations in transplantation patients.49 Tolerance at lower doses is comparable to that of acyclovir.
Localized bullous skin lesions50 and an acute generalized pustulosis confirmed by patch testing have been reported.51
Acyclovir has shown mutagenic activity in some in vitro assays at high concentrations, but no significant immunosuppressive activity, carcinogenicity, or teratogenicity has been noted in animal studies. High doses decrease spermatogenesis and cause testicular atrophy in animals. Acyclovir is classified as pregnancy category B and is present in breast milk. No excess frequency of congenital abnormalities has been recognized in infants born to women exposed to acyclovir during pregnancy, although whether exposure may increase the risk for spontaneous abortion is unresolved.52,53,54
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


