Anemias Secondary to Chronic Disease and Systemic Disorders
Robert T. Means, Jr.
ANEMIA OF CHRONIC DISEASE
The anemia that is often observed in patients with infectious, inflammatory, or neoplastic diseases that persist for more than 1 or 2 months is called anemia of chronic disease (ACD). The characteristic feature of this syndrome is the occurrence of hypoferremia in the presence of ample reticuloendothelial iron stores. ACD is defined by the presence of this unique combination of findings.1, 2, 3 As so defined, the syndrome does not include anemias caused by marrow replacement, blood loss, hemolysis, renal insufficiency, hepatic disease, or endocrinopathy, even when those disorders are chronic. These other syndromes are discussed in the sections “Anemia of Chronic Renal Insufficiency,” “Anemia in Cirrhosis and Other Liver Diseases,” and “Anemias Associated with Endocrine Disorders” in this chapter. To further complicate nomenclature, the acute anemia observed in critically ill patients and that component of postsurgical anemia not attributable to blood loss appear to be pathophysiologically identical to ACD.
As the discussion above suggests, the designation ACD is far from perfect.4 The alternate term, anemia of inflammation has become more widely used in recent years,4, 5 but also has significant etymologic deficiencies, and the more pathophysiologically correct term cytokine-mediated anemia6 is not commonly used. Highly specific descriptive designations, such as anemia of defective iron reutilization,7 hypoferremic anemia with reticuloendothelial siderosis, and thesauric hypoferremic anemia,8 are also rarely used and have the limitation of focusing solely on the iron-related aspects of the syndrome.
Associated Syndromes
ACD is extremely common and, overall, is probably more common than any anemia syndrome other than blood loss with consequent iron deficiency. Cash and Sears evaluated all the anemic individuals admitted to the medical service of a busy municipal hospital during two 2-month periods in 1985 and 1986.9 After patients with active bleeding, hemolysis, or known hematologic malignancy were excluded, 52% of anemic patients met laboratory criteria for ACD.9 The syndrome is also observed in 27% of outpatients with rheumatoid arthritis10 and in 58% of new admissions to inpatient rheumatology units.11 However, it should be remembered that 40% of patients in the series reported by Cash and Sears lacked one of the traditional “ACD-associated disorders.”9 Approximately one-third of this latter group had renal insufficiency, in which pathophysiologic mechanisms implicated in ACD are active.12 Clinical disorders commonly associated with ACD are listed in Table 41.1.
Clinical and Laboratory Description
Because this type of anemia occurs in association with so many diseases, the clinical manifestations necessarily vary widely. Usually, the signs and symptoms of the underlying disorder overshadow those of the anemia, but on rare occasions, developing anemia provides the first evidence of the primary condition. This situation may be observed particularly in difficult-to-diagnose clinical syndromes, such as temporal arteritis.13
Anemia
Development and Severity
Typically, anemia develops during the first 1 to 2 months of illness and thereafter does not progress.2 The hematocrit usually is maintained between 0.25 and 0.40,2, 14, 15 but significantly lower values are observed in 20% to 30% of patients.9, 10 The hemoglobin concentration and hematocrit generally provide an accurate reflection of the extent to which the circulating red cell mass is reduced, although in certain cases, expansion of the total blood volume would mean that the reduction in red cell mass is less than the hemoglobin or hematocrit indicates.16, 17
A general correlation exists between the degree of anemia and the severity of the underlying disease.2 For example, infections accompanied by pronounced fever, chills, and suppuration are associated with more severe anemia than those with fewer systemic manifestations.18 In infected wounds, the degree of anemia
is related to the number of organisms present.18 Correlation has also been observed between the severity of the anemia and the activity of rheumatoid arthritis as judged by fever, severity of joint swelling and inflammation, and the erythrocyte sedimentation rate.19, 20 In patients with malignant disease, anemia is more severe when metastases are widespread than when the disease is localized; however, the development of anemia does not require or imply neoplastic involvement of the bone marrow.21, 22
is related to the number of organisms present.18 Correlation has also been observed between the severity of the anemia and the activity of rheumatoid arthritis as judged by fever, severity of joint swelling and inflammation, and the erythrocyte sedimentation rate.19, 20 In patients with malignant disease, anemia is more severe when metastases are widespread than when the disease is localized; however, the development of anemia does not require or imply neoplastic involvement of the bone marrow.21, 22
TABLE 41.1 CONDITIONS ASSOCIATED WITH ANEMIA OF CHRONIC DISEASE | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Morphologic Features
The erythrocytes usually are normocytic and normochromic; however, hypochromia and microcytosis may be observed. In older series, microcytosis (mean corpuscular volume [MCV] <80 fl) was observed in 2% to 8% of patients with ACD19, 23, 24; however, other, more recent studies report a frequency of 20% to 40%.9, 15 Hypochromia (mean corpuscular hemoglobin concentration, 26 to 32 g/dl) is more common than microcytosis. In various series, hypochromia was observed in 23% to 50% of patients with chronic infection, 50% to 100% of patients with rheumatoid arthritis, and 44% to 64% of patients with cancer.2, 15 Overall, it is observed in 40% to 70% of patients with ACD.9, 10, 19, 23, 24 Hypochromia may be observed even though the hematocrit remains within normal limits.25 Microcytosis in ACD is usually not as striking as that commonly associated with iron deficiency anemia; values for MCV <72 fl are rare.2, 15 Another distinction from iron deficiency is that hypochromia typically precedes microcytosis in ACD but typically follows the development of microcytosis in iron deficiency.2 Slight anisocytosis and poikilocytosis may be detected, but such changes tend to be less prominent than in iron-deficient subjects. Routine examination of the blood smear rarely reveals specific morphologic abnormalities. The width of the erythrocyte size distribution curve (red cell distribution width) is typically elevated to a moderate degree, and generally does not help in distinguishing iron deficiency and ACD. Newer parameters calculated by automated hematologic analyzers, such as reticulocyte hemoglobin content (CHr), may assist in distinguishing ACD from iron deficiency.26, 27
Laboratory Markers of Iron Status
Characteristically, serum iron concentration is decreased, total iron-binding capacity (or serum transferrin concentration) is reduced, and transferrin saturation may be below normal, though not to the same degree as in iron deficiency.2, 24 In patients with infection, hypoferremia develops early in the course of the illness, often within 24 hours, and is observed even in acute, self-limited febrile diseases or after experimentally induced fever in humans or animals.8, 14, 28, 29 When the infection is of short duration, the serum iron returns to normal and anemia does not develop; in prolonged illnesses, the serum iron level remains low as long as the disease is active. When the disorder subsides, anemia often is relieved before the serum iron level returns to normal. The degree of hypoferremia is related to the severity of the underlying illness.19, 24, 30
In bone marrow aspirates stained for iron, the number of sideroblasts is reduced to 5% to 20% of the total quantity of normoblasts (normal, 30% to 50%). In contrast, the amount of hemosiderin within macrophages usually is increased; exceptions to this probably represent cases complicated by iron deficiency.2
Serum ferritin level is a useful indicator of iron status in patients without underlying inflammatory disorders. In patients with ACD, however, the serum ferritin level indicative of adequate reticuloendothelial iron stores requires upward adjustment. Serum ferritin values usually increase in patients with inflammatory diseases,31 and extreme elevations of serum ferritin may be a nonspecific indicator of significant underlying disease.32
When iron deficiency coexists, the serum ferritin level falls but may not reach values as low as those found in uncomplicated iron deficiency. Values of 60 to 100 µg/L, previously suggested as the appropriate lower limit of normal for serum ferritin in chronic inflammation, may be too low, depending upon the parameters of the ferritin assay used.33, 34, 35, 36 At one institution, all patients with serum ferritin levels <30 µg/L were iron-deficient by marrow examination, as were the majority of hospitalized patients with serum ferritin levels of 30 to 100 µg/L and approximately one-third of hospitalized patients with serum ferritin levels between 100 and 200 µg/L (Fig. 41.1).37 Combining the serum ferritin level with other parameters, such as erythrocyte sedimentation rate and C-reactive protein, did not improve its predictive value,38 although it has been suggested that the combination of serum ferritin with red cell ferritin may be more predictive.39 The recognition of iron deficiency in patients with chronic inflammatory states is not a trivial issue: Iron deficiency contributes to anemia in up to 27% of anemic rheumatoid arthritis patients10 and probably accounts for periodic reports of successful treatment of “the anemia of chronic disease” with iron preparations.40 A patient with chronic inflammatory disease and a serum ferritin <30 µg/L is certainly iron-deficient, and a patient with a serum ferritin >200 µg/L is certainly not iron-deficient; in other circumstances, certainty can be provided only by examination of a Prussian blue-stained marrow specimen. Elevated concentration of soluble transferrin receptors (sTfRs) in serum is another way to identify iron-deficient individuals with normal serum ferritin concentrations.41
Serum or plasma sTfR concentration is elevated in iron deficiency but is not elevated in uncomplicated ACD. It will be elevated in ACD cases complicated by iron deficiency.42, 43, 44, 45 sTfR concentration is most accurately interpreted in the context of the serum ferritin concentration,41, 46 and is often expressed as a ratio to the log of the serum or plasma ferritin concentration (called the transferrin receptor index or transferrin/ferritin index).46
As will be discussed later in this chapter, the iron regulatory peptide hepcidin appears to be a significant driver of the pathogenesis of ACD. Circulating hepcidin concentration is expected to be elevated in ACD.5, 47 Well-studied and reproducible assays for hepcidin have been developed,48, 49, 50, 51, 52, 53 and while none are available for routine clinical use at present it is reasonable to assume that they will be available in the near future. It has been suggested that serum hepcidin concentration can be plotted against CHr and that the combination of elevated hepcidin and normal CHr will distinguish ACD from both iron deficiency (normal hepcidin, high CHr) and the combined state of ACD and iron deficiency (low CHr, high hepcidin).54
 FIGURE 41.1. Comparison of serum ferritin levels from iron-deficient and non-irondeficient patients as identified by bone marrow examination during 1994 to 1995 at the University of Cincinnati Medical Center. Dashed lines indicate the range of normal for the laboratory. |
Other Biochemical Findings
The concentration of free protoporphyrin in the erythrocytes (FEP) tends to be elevated in patients with ACD.14, 55, 56 However, FEP increases more slowly in ACD than it does in iron deficiency, and it does not become clearly abnormal until significant anemia has developed.
A variety of other biochemical changes often are detected in patients with chronic diseases. Many of these changes reflect alteration of levels of particular plasma proteins, often called acute-phase reactants.57, 58, 59, 60, 61 The concentrations of certain plasma proteins, such as fibrinogen, ceruloplasmin, haptoglobin, C-reactive protein, orosomucoid, C3, and amyloid A protein, increase62, 63, 64, whereas the concentrations of albumin and transferrin characteristically decrease.65 The increase in ceruloplasmin accounts for the increase in serum copper levels often noted in association with chronic diseases.2, 14 An elevated fibrinogen level is probably the most important factor in the increased sedimentation rate.
Patients with chronic illness develop accelerated protein catabolism and negative nitrogen balance associated with muscle proteolysis.64, 65 Over time, this phenomenon can result in muscle wasting, increased urea excretion, weight loss, and growth impairment in children. However, protein catabolism generates amino acids that can be used by the patient as alternative energy sources or to supply substrates for biosynthetic processes related to host response. In this context, it is perhaps relevant that elevated serum levels of tumor necrosis factor (TNF), a cytokine implicated in the pathogenesis of ACD, are noted in patients with significant malnutrition.66, 67
Kinetic Characteristics
Erythrocyte survival is modestly but significantly reduced in patients with ACD. In two studies comparing red cell survival in anemic patients with rheumatoid arthritis to red cell survival in normal individuals, the mean red cell survivals noted were 81 days versus 98 days, and 90 days versus 114 days, respectively.68, 69 In a similar study comparing red cell survivals in 10 anemic patients with a variety of chronic inflammatory states to 10 normal individuals, the mean values observed were 80 days versus 88 days, respectively.68 The usual manifestations of increased blood destruction, such as increases in serum bilirubin values and urobilinogen excretion, are not typically observed.2, 70
There is little evidence of a compensatory erythropoietic response to this reduction in red cell survival. The reticulocyte count usually is normal or decreased, and little or no erythroid hyperplasia of the marrow is observed. The pathogenetic significance of these findings is discussed in the following section. Kinetic data indicate that anemia develops because the bone marrow fails to increase red cell production sufficiently to compensate for a mild decrease in the lifespan of the red cells.2, 8, 68 Ferrokinetic studies involving patients with chronic infections,71, 72 rheumatoid arthritis,73 and various malignant diseases21, 22, 74 reveal that the rate of disappearance of iron from the plasma is rapid, the plasma iron transport rate is normal or slightly increased, the uptake of iron into erythrocytes and the amount of iron turning over through red cells daily are normal or increased, and the fraction of red cells renewed daily is increased.2 When techniques that allow the division of marrow iron turnover (a measure of total erythropoiesis) into red cell iron turnover and ineffective iron turnover were used, marrow iron turnover was normal in patients with ACD.68, 69, 75, 76 Ineffective iron turnover was also normal, or even less than normal, indicating a lack of ineffective erythropoiesis as traditionally defined.68 In iron-deficient subjects, ineffective iron turnover is increased, perhaps because of greater stimulation of the marrow.
Pathogenesis
Efforts to clarify the pathogenesis of ACD have focused on three principal abnormalities: shortened erythrocyte survival, impaired marrow response, and disturbance in iron metabolism. The modest shortening of the erythrocyte survival creates an increased demand for red cell production on the marrow. Normally, the marrow could easily accommodate this demand, but in the setting of ACD, the marrow is unable to respond fully because of a combination of a blunted erythropoietin (Epo) response, an inadequate progenitor response to Epo, and limited iron availability (Fig. 41.2).
Cytokines
ACD is one manifestation of the systemic response to immunologic or inflammatory stress, which results in the production of various cytokines.3, 77 The ability to trigger this cytokine response appears to be the common pathogenetic factor shared by the various conditions associated with this anemia syndrome.78 The central role of these molecules suggests that ACD may be best understood as a cytokine-mediated process.6 The cytokines most often implicated in the pathogenesis of ACD are TNF,79, 80, 81 IL-1,82, 83 IL-6,84 and the interferons,85, 86, 87 concentrations of which have been reported to be increased in the serum or plasma of patients with disorders associated with ACD.85, 87, 88, 89, 90, 91 Therapeutic administration of TNF or interferon may induce anemia.92, 93 The role of IL-6 is complex. IL-6 administration itself does not suppress erythropoiesis but rather is associated with increased plasma volume; therefore there is a dilutional component to any observed anemia.17 The association of IL-6 with hepatocyte activation may thus explain the dilutional component of the anemia observed in liver disease.94, 95, 96 However, IL-6 is also a potent inducer of hepcidin, proposed as a major mediator of the iron abnormalities of ACD.5
The Role of Hepcidin
While inflammation-induced cytokine activation is clearly the initial event in the pathogenesis of ACD, the liver-produced antimicrobial peptide hepcidin appears to be the most important mediator through which cytokines exert their effects on the pathogenetic mechanisms of ACD.5, 97, 98 Hepcidin is an acute-phase-reacting peptide, largely regulated by IL-6 but also by cytokine pathways not linked to IL-6.5, 99, 100 Patients with hepatic adenomas secreting hepcidin exhibit a hypoferremic anemia that resolves with resection of the adenoma.97 Urinary hepcidin excretion is strongly correlated with serum ferritin concentration and is markedly elevated in patients with anemia of inflammation (defined as anemia
with a characteristic clinical setting and an elevated serum ferritin) compared to iron-deficient patients.5 Hepcidin promotes macrophage iron retention by causing internalization of the iron export protein ferroportin.101
with a characteristic clinical setting and an elevated serum ferritin) compared to iron-deficient patients.5 Hepcidin promotes macrophage iron retention by causing internalization of the iron export protein ferroportin.101
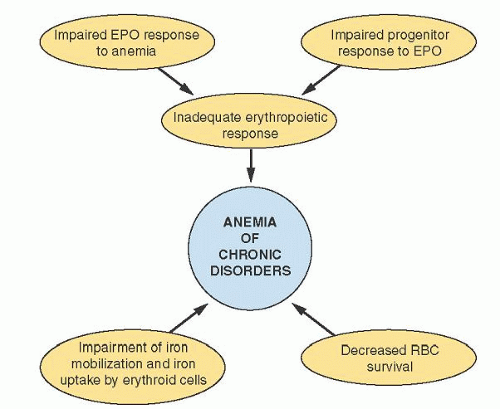 FIGURE 41.2. Schematic diagram representing contributing mechanisms in the pathogenesis of anemia of chronic disease. EPO, erythropoietin; RBC, red blood cell. |
Of the pathogenetic mechanisms shown in Figure 41.2, hepcidin clearly drives the abnormalities in iron metabolism. However, it may be linked to other elements as well. Under conditions of limited Epo availability, hepcidin is associated with impaired erythroid colony formation in vitro.102 Increased circulating hepcidin appears to be linked to the degree of resistance to recombinant (rh) Epo therapy in dialysis patients.103 Hepcidin and Epo production appear to be regulated in an inverse relationship by hypoxiainducible factor,104 which may potentially provide some linkage between hepcidin and the relative Epo deficiency of ACD. This latter connection remains to be demonstrated.
Shortened Erythrocyte Survival
The rate of survival of cells from patients with arthritis, when transfused into normal subjects, is normal, and the survival of red cells from normal individuals in the circulation of patients with arthritis is less than the normal rate.2, 23 Therefore, shortened red cell survival in patients with chronic inflammatory disorders is attributed to an extracorpuscular mechanism.30 IL-1 levels and shortened red cell survival are correlated in anemic patients with rheumatoid arthritis,105 and mice that become anemic after exposure to TNF in vivo also exhibit a shortened red cell survival.106 Neocytolysis, a selective hemolysis of newly formed erythrocytes associated with Epo deficiency,107, 108 has been proposed as a mechanism for shortened red blood cell survival in ACD. Peroxynitrite, derived from the reaction of the cytokine second messenger nitric oxide and superoxide, may contribute to red cell rigidity and thus to decreased survival.109
Impaired Marrow Response
Normal bone marrow, capable of a six- to eightfold increase in the red cell production rate, should easily compensate for such a modest reduction in erythrocyte survival. Its failure to do so in ACD suggests that impaired production capacity is of fundamental importance in the pathogenesis of this condition. The possible defects in erythropoiesis fall into three categories: inappropriately low Epo secretion, diminished marrow response to Epo, and ironlimited erythropoiesis.
An inverse relationship between serum or plasma Epo levels and hemoglobin normally exists: As the hemoglobin decreases, the Epo level rises.110 A similar inverse relationship between hemoglobin and Epo level exists in anemic individuals with rheumatoid arthritis, cancer, and human immunodeficiency virus infection111, 112, 113, 114; however, for any given anemic patient in these disease categories, the Epo level was lower than that found in equally anemic individuals with iron deficiency, indicating that the Epo response to anemia was blunted. This impaired Epo response is cytokine-mediated. IL-1, TNF-α, and transforming growth factorβ inhibit production of Epo by various hepatoma cell lines and by isolated perfused rat kidneys in vitro.115, 116 This effect occurs at the messenger RNA level.115 It has been proposed that this inadequate Epo secretion is adaptive; that is, it reflects reduced tissue oxygen use so that normal oxygenation is maintained despite reduced hemoglobin levels. One colorful description of this process is that the “hematologic thermostat has been turned down a bit”,8 perhaps to allow diversion of substrates normally used in erythropoiesis to more immediately critical activities. Changes that ordinarily signify erythrocyte adaptation to tissue hypoxia, such as increased erythrocyte 2,3-diphosphoglycerate (2,3-DPG) levels, are observed in ACD.117 Hemoglobin oxygen affinity is also slightly decreased in these patients but overlaps the range observed in normal subjects.117
Although the Epo levels of patients with ACD are lower than those observed in equally anemic iron-deficient individuals, they are still higher than are observed in normal individuals who are not anemic. This implies that inhibition of Epo production cannot entirely account for the impaired erythropoiesis associated with ACD, and that the erythroid progenitors themselves exhibit an abnormal response to Epo. Studies of anemic cancer patients support this concept.118, 119
TNF, IL-1, and interferon have all been reported to inhibit erythropoiesis in vivo and in vitro.90, 92, 120, 121, 122, 123, 124, 125, 126, 127, 128 TNF inhibits in vitro colony formation by human erythroid colony-forming units (CFU-E) indirectly, through interferon-β and possibly other soluble factors released from marrow accessory cells in response to TNF.129, 130 The inhibitory effect of rhIL-1 on human CFU-E colony formation was also indirect and dependent on marrow accessory cells; however, the responsible accessory cells in this case were T lymphocytes, and the mediator was interferon-γ, which also exhibits a direct inhibitory effect on human CFU-E.131 Although interferon-α and interferon-β share a common receptor, interferon-α does not exert a direct inhibitory effect on human CFU-E colony formation. Instead, its effect is mediated through an unidentified soluble factor released by T lymphocytes.132 The inhibitory effects of interferon-α and interferon-β exhibit synergy with interferon-γ.132 An important point to consider in reviewing the various models for cytokine effects on erythropoiesis is that none of the systems described operates in isolation in vivo. Thus, TNF induces IL-1 production by macrophages, IL-1 induces interferon-γ production by T lymphocytes, and interferon-γ can exhibit positive or negative feedback on production of IL-1 and TNF.
Abnormal Iron Metabolism
It has been proposed that lack of iron for erythropoiesis contributes to the inadequate marrow response in ACD. Evidence of a functional iron deficiency in this syndrome includes erythrocyte microcytosis, increased FEP,14, 56 reduced transferrin saturation,24 and decreased marrow sideroblasts.24
Ferrokinetic studies have proved confusing. It has been reported that erythroid iron turnover correlated with serum iron levels, permitting a conclusion that marrow proliferation was limited by availability of iron in ACD.118 Other studies found no correlation between marrow iron turnover and serum iron concentration.68, 69, 76, 136
The major contributor to hypoferremia in patients with ACD is probably a shift of iron from a transferrin-bound, available circulating state to an intracellular storage state. Iron absorption appears to be normal, but iron tends to remain in the mucosal cell and in hepatocytes.137, 138 Macrophages, the major site from which iron is obtained for erythropoiesis, also exhibit increased iron storage. This process is mediated by hepcidin, which downregulates the iron egress regulatory ferroportin, thus trapping iron intracellularly.101
The dominant factor in the iron abnormalities of ACD is clearly hepcidin, discussed above and at greater length in Chapter 23. A number of other processes of less overarching significance may contribute under particular circumstances to the iron anomalies observed. Apoferritin is normally synthesized in response to increased intracellular iron concentration.139 It has been suggested that excess apoferritin is made in inflammatory and malignant conditions, and the surplus binds a larger-than-usual amount of iron entering the cell.140, 141, 142 In effect, such a mechanism would divert iron from the rapid to the slow pathway of iron release. Rodents injected with recombinant TNF developed
a hypoferremic anemia associated with impaired storage iron release and incorporation into erythrocytes.106, 143 It has also been reported that IL-1 increases translation of ferritin messenger RNA and that this additional ferritin could act as a trap for iron that might otherwise be available for erythropoiesis.105 Nitric oxide, which is a common mediator of cytokine effects, has similar effects on ferritin expression.144, 145 Lactoferrin is a transferrinlike protein found in neutrophil-specific granules.146, 147 It is released from the neutrophil during phagocytosis or stimulation by IL-1.148 At low pH, lactoferrin binds iron more avidly than does transferrin.149 Lactoferrin-bound iron is not immediately available for erythropoiesis150; rather, it binds to specific receptors on macrophages (particularly in the liver and spleen) and is endocytosed and subsequently incorporated into ferritin.151, 152 Thus, lactoferrin transfers iron from its transferrin-bound, circulating state to a storage state, from which it cannot be rapidly mobilized.153 One group of investigators found that reducing the available lactoferrin by inducing neutropenia in rats blunted the hypoferremic response to IL-1,154 but another group found that administration of recombinant IL-1-induced hypoferremia in mice, even in the face of severe neutropenia.155
a hypoferremic anemia associated with impaired storage iron release and incorporation into erythrocytes.106, 143 It has also been reported that IL-1 increases translation of ferritin messenger RNA and that this additional ferritin could act as a trap for iron that might otherwise be available for erythropoiesis.105 Nitric oxide, which is a common mediator of cytokine effects, has similar effects on ferritin expression.144, 145 Lactoferrin is a transferrinlike protein found in neutrophil-specific granules.146, 147 It is released from the neutrophil during phagocytosis or stimulation by IL-1.148 At low pH, lactoferrin binds iron more avidly than does transferrin.149 Lactoferrin-bound iron is not immediately available for erythropoiesis150; rather, it binds to specific receptors on macrophages (particularly in the liver and spleen) and is endocytosed and subsequently incorporated into ferritin.151, 152 Thus, lactoferrin transfers iron from its transferrin-bound, circulating state to a storage state, from which it cannot be rapidly mobilized.153 One group of investigators found that reducing the available lactoferrin by inducing neutropenia in rats blunted the hypoferremic response to IL-1,154 but another group found that administration of recombinant IL-1-induced hypoferremia in mice, even in the face of severe neutropenia.155
In addition to decreased availability of iron, erythroid progenitors may also be unable to fully use the iron available to them. Erythroblasts from anemic patients with rheumatoid arthritis express fewer surface TfR than do erythroblasts from normal individuals. These TfR also exhibit lower binding affinity for transferrin.156 Furthermore, acute-phase reactants, such as α1-antitrypsin, impair transferrin binding to erythroblasts and also inhibit transferrin internalization.157 rhEpo appears to induce a greater level of TfR expression on erythroid cells.158
Anemia in Patients with Cancer
Much of the anemia commonly observed in patients with cancer can be attributed to the mechanisms involved in ACD; however, certain processes unique to malignancy may also contribute. Erythroid precursors may be displaced from marrow by metastatic tumor,159 tumor-induced fibrosis,160 or tumor-associated marrow necrosis.161 The treatment of cancer can also produce or exacerbate anemia by a variety of mechanisms, including impaired Epo production162 and cytotoxic effects of therapy on erythroid progenitors.163
Diagnosis
Studies suggest that diagnosis of anemia of moderate degree, as is commonly observed in ACD, is often missed.164 ACD should be considered in anemic patients with associated inflammatory, infectious, or neoplastic states. As described in Table 41.1,9 not all cases are associated with a classic chronic disease, but virtually all cases are associated with states of cytokine or immune activation. This is likely the mechanism associated with the increasing number of reports of ACD in congestive heart failure.103, 165, 166, 167 The diagnosis is confirmed by demonstrating hypoferremia with adequate reticuloendothelial iron stores in a patient with an appropriate clinical syndrome. Typically, the serum transferrin is either low or low normal, and sTfR concentration is normal in ACD. The major differential diagnosis is iron deficiency anemia. This is not a trivial distinction. The diagnosis of iron deficiency mandates identification of a source of blood loss. Incorrectly labeling a patient with ACD as iron-deficient exposes that patient to intrusive and expensive (although fairly safe) diagnostic procedures and to ineffective therapy. Mislabeling an iron-deficient patient as having ACD may result in failure to diagnose an underlying gastrointestinal malignancy at a curable stage and in failure to offer inexpensive and effective therapy. The diagnosis of iron deficiency is discussed in detail in Chapter 23 and in the section “Abnormal Iron Metabolism.” It has been suggested that a marker of inflammation or cytokine activation, such as C-reactive protein or IL-6 should be an element of the diagnosis of ACD.168,169 This may not be necessary in cases of ACD associated with “classic” ACD-related disease, like rheumatoid arthritis, but may be helpful in ACD which are less typical, or perhaps complicated by iron deficiency.
In principle, absence of an elevated serum or plasma hepcidin concentration could rule out ACD. When hepcidin assays become available for routine use in a clinical context, it will be important to carry out well-designed studies to determine whether they will contribute more to diagnosis than currently available clinical markers of iron status.170
Treatment
The focus of therapy should be on the underlying disorder. The anemia itself is rarely an important clinical problem. Thus, direct approaches to correction of the anemia are rarely necessary. Fewer than 30% of patients have anemia sufficiently severe to necessitate transfusion, and assessment of the symptomatic state should always be considered before administration of blood products.
rhEpo and the rhEpo analog darbepoetin, are effective in ACD but expensive,171, 172, 173 and many if not most third-party payers will not reimburse for its use in ACD. It is not currently approved for this purpose in the United States. Limitations on the use of rhEpo in anemic cancer patients for safety reasons (discussed below) limit the use of rhEpo in ACD at present. Because most patients were not symptomatic from their anemia, symptomatic benefit was rarely reported in the initial studies.171, 172 Subsequent studies using more sophisticated evaluation instruments have reported increased quality of life in anemic patients with rheumatoid arthritis174 or cancer175 treated with Epo. It can also be used for ACD patients who wish to donate blood for autologous transfusion at elective surgery but are too anemic to do so176 or to permit autologous blood donation by a patient with ACD and multiple alloantibodies.177 It has been proposed that rhEpo administration may be of benefit in anemic patients with congestive heart failure.178, 179, 180, 181, 182
It is debated whether or not to administer iron routinely to patients receiving therapy with Epo products. It is this author’s practice to do so in the absence of elevated serum ferritin levels. In one study of anemic patients with rheumatoid arthritis, the concurrent use of iron supplementation was a powerful predictor of response to Epo183; however, many of the patients in this study may have been iron-deficient. Although there are reports of correction of ACD by intravenous iron without Epo,40, 184 normalization of hemoglobin was largely seen in patients who were also iron-deficient.40 Iron therapy by itself is likely to be useful only in patients who have concurrent iron deficiency,10 and then only for the component of anemia caused by iron deficiency. There is no convincing evidence that iron alone corrects ACD per se. Anemic cancer patients treated with concurrent intravenous iron and rhEpo appear to have a better response than those treated with no iron supplementation or with iron supplementation alone.185
Studies of the use of rhEpo in cancer patients have been associated with increased adverse outcomes in certain cases,186, 187, 188, 189, 190 leading to restrictions on circumstances in which rhEpo can be used in cancer therapy. While there is debate in the literature as to whether the observed outcomes reflected unique features of particular rhEpo regimens or of specific patient populations,191, 192, 193 the clinician is encouraged to review current guidelines prior to initiating therapy.
As noted earlier, Epo downregulates hepcidin production. Given the significant role of hepcidin in the pathogenesis of ACD, therapy directed against hepcidin would be attractive in principle. Antisense hepcidin has been reported to decrease anemia in an animal inflammation model.194 At present, however, there are no specific antihepcidin agents available for clinical use.
ANEMIA OF CHRONIC RENAL INSUFFICIENCY
Anemia is an almost invariable manifestation of chronic renal failure, often contributing substantially to the morbidity of the condition. It may be considered as typical of the disease as azotemia.195 In the era before the availability of rhEpo, 98% of patients on hemodialysis were anemic196; even now, 48% of renal failure patients before dialysis and 28% of hemodialysis patients have hematocrit values <0.30.197,198 The term anemia of chronic renal insufficiency refers to that anemia resulting directly from failure of the endocrine and filtering functions of the kidney. The kidney is the major source of Epo, and the ability to secrete this hormone is lost as the kidney fails. In addition, renal failure is associated with other pathologic processes, including some that may inhibit erythropoiesis and others that may shorten erythrocyte survival. Lack of sufficient Epo is by far the most important of these anemia-causing factors; consequently, the hypoproliferative features of the anemia tend to predominate.199
In clinical settings associated with chronic renal failure, additional factors may also contribute to the development of anemia, but these should be considered complications rather than fundamental components of the anemia of renal insufficiency itself. In the presence of infection or inflammation, ACD is likely to be observed. Iron deficiency anemia (see Chapter 23) may develop because of blood loss from the gastrointestinal tract or (less frequently) hematuria or from retention of blood in the hemodialysis apparatus tubing.199, 200 The megaloblastic anemia of folate deficiency also may occur in patients on dialysis201, 202 but is otherwise uncommon.203 Certain types of renal disease, including the hemolytic-uremic syndrome or thrombotic thrombocytopenic purpura, are associated with microangiopathic hemolytic anemia (see Chapter 48). Finally, aluminum intoxication can cause microcytic anemia in dialysis patients, although this has become rare in modern dialysis practice.204
Clinical Description
Chronic renal failure occurs during the final stages of several renal diseases. As a rule, the nature of the underlying disease bears little relation to the degree of anemia, although anemia may be less severe in patients with hypertensive renal disease205 and is considerably less severe in patients with polycystic disease.206, 207 In one series, the mean hematocrit in 12 subjects with polycystic disease was 0.297, compared with 0.212 in 24 subjects with other types of chronic renal failure.207 Apparently, the Epo-secreting function of the kidney is preserved in polycystic disease—even when the filtering function is lost.207, 208 Erythropoietic activity can be found in the cystic fluid and may arise from single interstitial cells juxtaposed to proximal tubular cysts.207
In most instances, the patient seeks medical attention because of symptoms related to the underlying renal disease, and anemia is an incidental finding. Occasionally, however, the renal symptoms are so subtle and so slowly progressive that the patient cites only symptoms of pallor, exertional dyspnea, or other signs of the cardiovascular adjustment to anemia. The severity of the anemia bears a rough relationship to the degree of renal insufficiency. Anemia is not routinely observed until the creatinine clearance falls to <45 ml/minute/1.73 m2 body surface area, which corresponds roughly to a serum creatinine of 2.0 to 2.5 mg/ml in an average-sized adult.209 At creatinine clearance rates below that, a statistically significant correlation between creatinine clearance and hematocrit has been reported.210, 211 However, the variation in the results of these studies is so great that one cannot reliably predict the hemoglobin level in an individual patient on the basis of renal function.
Laboratory Findings
Anemia tends to become more severe as renal failure worsens, but in most patients the hematocrit ultimately stabilizes between 0.15 and 0.30.2 Because regulation of body water and electrolyte balance is impaired in renal disease, the apparent degree of anemia may be exaggerated or minimized by alterations in plasma volume.
The erythrocytes usually are normocytic and normochromic, but slight macrocytosis is occasionally observed.212 The majority of red cells appear normal on blood smears. Occasionally, however, “burr” cells (Fig. 41.3) are observed along with some triangular, helmet-shaped, or fragmented cells. The reticulocyte count often is within normal limits,213 but it may be moderately increased.212, 214 In one study, the numbers of reticulocytes were normal when the blood urea nitrogen (BUN) value was <130 mg/dl; at higher BUN levels, however, their number often was increased.215 The highest values (average, 6%) were observed with extreme azotemia (BUN, 300 to 350 mg/dl).
The leukocyte count typically is normal, but slight neutrophilic leukocytosis may be observed. In one series, the leukocyte count averaged 10.7 × 109/L.213 The platelet count is either normal or slightly increased,213 but platelet function may be severely impaired, resulting in defective hemostasis (see Chapter 52).
Related posts:
 The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias During Pregnancy and The Postpartum Period
The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias During Pregnancy and The Postpartum Period
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


