INTRODUCTION
SUMMARY
Most patients suffering from chronic infection, chronic inflammation, or some with various malignancies develop a mild to moderate anemia. This anemia, designated anemia of chronic disease or anemia of chronic inflammation, is characterized by a low serum iron level, a low to normal transferrin level, and a high to normal ferritin level. The anemia is caused by the direct and indirect inhibitory effects of inflammatory cytokines on erythrocyte production. Among the cytokines, interleukin-6 has a central role, acting by increasing hepatocyte production of the iron-regulatory hormone hepcidin. Hepcidin then blocks the release of iron from macrophages and hepatocytes, causing the characteristic hypoferremia associated with this anemia, and limiting the availability of iron to the developing erythrocytes. Effective treatment of the underlying disease restores normal erythropoiesis. When this is not possible, and treatment is necessary, therapeutic trials have revealed that the anemia is often responsive to pharmacologic doses of erythropoietin.
Anemia of chronic kidney disease presents similarly to anemia of inflammation but because the kidneys are the predominant site of erythropoietin production, the pathogenesis of this anemia is frequently dominated by relative erythropoietin deficiency, where erythropoietin concentrations in serum are lower than expected for the severity of anemia. Systemic inflammation from underlying renal disease, or induced by dialysis treatments and their complications, contributes to pathogenesis in a manner similar to anemia of inflammation. Circulating hepcidin concentrations may also rise because of its decreased renal clearance. Suppressive effects of uremia on erythropoiesis and blood losses from hemodialysis may contribute to anemia in end-stage renal disease. A combination of erythropoiesis–stimulating agents and intravenous iron is usually effective in reversing anemia but overtreatment may worsen overall outcomes.
Acronyms and Abbreviations:
ACD, anemia of chronic disease; AI, anemia of inflammation; CKD, chronic kidney disease; CPG, clinical practice guideline; CRP, C-reactive protein; EPO, erythropoietin; ESA, erythropoiesis-stimulating agent; IDA, iron-deficiency anemia; IL, Interleukin; KDIGO, The Kidney Disease Improving Global Outcomes; sTfR, soluble transferrin receptor; TfR, transferring receptor; TNF, tumor necrosis factor.
DEFINITION AND HISTORY
The term anemia of chronic disease (ACD) or anemia of chronic disorders refers to mild to moderately severe anemia (hemoglobin [Hgb] 7 to 12 g/dL) associated with chronic infections and inflammatory disorders and some malignancies.1 The newer name, anemia of inflammation (AI), is not only more reflective of the pathophysiology of ACD but also includes anemia of critical illness,2 a condition that presents similarly to ACD but develops within days of the onset of illness. An anemia similar to AI is seen in some older individuals in the absence of an identifiable chronic disease; this condition is sometimes referred to as unexplained anemia of elderlies or anemia of aging (Chap. 9).3
AI is characterized by inadequate erythrocyte production in the setting of low serum iron and low iron-binding capacity (i.e., low transferrin) despite preserved or even increased macrophage iron stores in the marrow. The erythrocytes are usually normocytic and normochromic but can be mildly hypochromic and microcytic. Anemia of critical illness2 can develop acutely (within days) in intensive care settings where the effects of infection or inflammation are exacerbated by disease-related or iatrogenic blood loss or red cell destruction, which by themselves are not sufficiently severe to cause anemia. Anemia of aging3 is diagnosed in the older when a normocytic normochromic anemia with low serum iron and preserved iron stores develops without an identified underlying disease. Older patients in this defined subset typically have an elevated sedimentation rate and/or elevated C-reactive protein (CRP), a high plasma interleukin (IL)-6 concentration, and frailty.
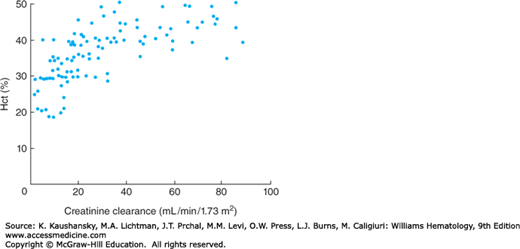
Anemia of chronic kidney disease (CKD) usually develops as chronic renal disease progresses and generally becomes more severe with decreasing creatinine clearance (Fig. 37–1). The anemia presents similarly to AI but because the kidney is the main site of erythropoietin (EPO) production in adults, the progressive destruction and fibrosis of the kidneys causes relative EPO deficiency, which frequently dominates the pathogenesis of this anemia. Patients with polycystic kidney disease are often at least partially spared of the anemia, whereas patients with bilateral nephrectomy are particularly severely affected by EPO deficiency. Systemic inflammation, true iron deficiency and decreased clearance of hepcidin are common consequences of the underlying disease and dialysis treatments, and one or more of these factors frequently worsen anemia or diminish the response to EPO therapy.
Figure 37–1.
Relationship between hematocrit (Hct) and creatinine clearance in patients with CKD. Anemia worsens with decreasing creatinine clearance. (Reproduced with permission from Radtke HW, Claussner A, Erbes PM, et al: Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function Blood 1979 Oct;54(4):877-884.)
Physicians have known about the pale appearance of patients with chronic infections for hundreds of years. In 19th-century Europe, tuberculosis was the major killer and the pallor associated with this disease was romanticized in the art literature of the time. The first measurements of red cell mass revealed the association between inflammation and anemia. Discussing “the alterations in the condition of the Blood in Inflammation” in Section 372 of the 1859 edition of the Principles of Human Physiology, William B. Carpenter4 described this connection between inflammation and anemia (author’s parentheses): “With this increase in the proportion of fibrin and colorless corpuscles (leukocytes), separately or in combination, there is a diminution of the proportion of the red corpuscles, albumen and the salts of the blood.” In 1961, 100 years later, Maxwell Wintrobe, in the fifth edition of Clinical Hematology,5 used the term “simple chronic anemia” for the normocytic anemia associated with the majority of infections and chronic systemic diseases. He described anemia associated with inflammation as a common subtype. Wintrobe proposed “profound alterations in iron and porphyrin metabolism” as the likely cause, and referred to his own experiments that showed a decrease in erythrocyte survival of only 27 percent, which “could easily be met by increased erythropoiesis if the bone marrow functional capacity were not impaired.” Despite advances in our understanding of the pathophysiology of this very common form of anemia, our knowledge is incomplete.
Anemia of CKD became a common problem in the 1960s when hemodialysis became widely available and allowed prolonged survival of patients with end-stage renal failure. Anemia of CKD was usually severe enough to limit activities of daily living and was treated by blood transfusions until the late 1980s when recombinant EPO became widely available, and alleviated the most severe forms of this anemia.
EPIDEMIOLOGY
The high prevalence of infectious diseases worldwide and the high prevalence of inflammatory and malignant disorders in industrialized countries would suggest that AI is the second or third most common form of anemia after iron-deficiency anemia (IDA) and possibly thalassemia.6 Although the prevalence of iron deficiency in the industrialized countries is now rapidly decreasing,6,7 AI is expected to increase as the population ages. Table 37–1 lists the most common diseases associated with AI.
| Category | Diseases Associated with Anemia of Inflammation |
|---|---|
| Infection | AIDS/HIV, tuberculosis, malaria (contributory), osteomyelitis, chronic abscesses, sepsis |
| Inflammation | Rheumatoid arthritis, other rheumatologic disorders, inflammatory bowel diseases, systemic inflammatory response syndrome |
| Malignancy | Carcinomas, multiple myeloma, lymphomas |
| Cytokine dysregulation | Anemia of aging |
Although anemia can develop early in the progression of CKD, it generally worsens as the kidneys fail.8,9,10 Accordingly, the prevalence of patients with anemia of CKD worldwide is influenced by the availability of life-sustaining dialysis therapies. It is estimated that there are currently approximately 600,000 patients with end-stage renal disease in the United States, and approximately 100,000 new patients each year,11 the majority of whom are anemic or receive treatment for anemia.9 Additional patients with milder anemia of CKD are found among the estimated 6.7 percent of the U.S. population (or approximately 20 million) identified as having likely CKD (estimated glomerular filtration rate [eGFR] <60 mL/min/1.73 m2) in the 2007–2010 National Health and Nutritional Examination Surveys (NHANES) study.11
ETIOLOGY AND PATHOGENESIS
In the chronic setting, AI predominantly results from the body’s inability to increase erythrocyte production to compensate for relatively small decrements in erythrocyte survival (reviewed in Ref. 1). In the steady state, erythrocyte production is sufficiently high so that the resulting anemia is mild to moderate. The anemia associated with acute critical illness has the same pathogenesis as other forms of AI but it develops more rapidly perhaps because of the more extensive erythrocyte destruction and intensive diagnostic phlebotomy common in this setting. The key questions about the pathogenesis of AI, still only partially answered, are: (1) What accounts for the inability of the AI marrow to increase erythropoiesis? and (2) How is this deficit connected to the characteristic hypoferremia and sequestration of iron in macrophages and hepatocytes? Anemia of CKD is similar to AI but the underlying renal pathology also impairs the ability of the kidneys to produce enough EPO leading to insufficient compensatory erythropoiesis.
Human studies indicate that transfused AI erythrocytes have a normal life span in normal recipients but transfused normal erythrocytes have a decreased life span in AI recipients.1 This finding suggests that increased erythrocyte destruction is caused by the activation of hosts factors such as macrophages that prematurely remove aging erythrocytes from the bloodstream. The explanation is consistent with the predominance of young erythrocytes in AI. Whether extrinsic factors, such as bacterial toxins and medications, or host-derived antibodies or complement contribute to this process is unknown.
Some cytokines, chiefly tumor necrosis factor (TNF)-α, IL-1, and the interferons, exert a suppressive effect on erythroid colony formation.12 Interferon-γ overproduction suppresses erythropoiesis in a mouse model13 by reducing erythrocyte life span and decreasing erythropoiesis without any evidence of iron restriction. It is not known to what extent and under what conditions these mechanisms contribute to human AI.
The normal response to increased destruction of erythrocytes is transient anemia followed by an increase in EPO production and subsequent compensatory increase in erythropoiesis. One proposed explanation for the inadequate marrow response in AI is less EPO production than expected based on other types of anemia. Studies of patients with rheumatoid arthritis and AI indicated that EPO levels are increased but less so than in IDA.14,15,16,17,18,19 The findings were similar in patients with anemias associated with solid tumors or hematologic malignancies.20,21 However, these comparisons did not take into account the potentiating effect of iron deficiency on hypoxia sensing (Chaps. 32 and 42).22 This effect could increase EPO production in IDA above that in other types of anemia, and make EPO production in AI appear low in comparison. In support of the EPO suppression hypothesis are experiments with EPO-producing cell lines indicate that production of the hormone is inhibited by inflammatory cytokines including TNF-α and IL-1. The inhibition is mediated by the effects of the transcription factor GATA-1 on the EPO gene promoter, and the suppression of EPO production can be reversed by a GATA inhibitor.23 Moreover, both baseline and hypoxia-induced EPO gene expression is suppressed in rats treated with bacterial lipopolysaccharide or IL-1β to mimic a septic state.24 However, suppression of EPO production is not the major mechanism of AI. If it were, administration of relatively small amounts of EPO should be sufficient to reverse the AI.
In contrast, relative EPO deficiency is often a major contributor to anemia of CKD. Most destructive diseases affecting the kidneys also decrease the release of EPO.25,26 In the kidney, interstitial fibroblasts of neural crest origin26,27 are probably the main source of EPO, but the identity of EPO-producing cells in the kidney remains controversial, mostly because the basal production of EPO is very low and ultrasensitive methods are required to detect the source of the hormone. In response to anemia or hypoxia, the number of renal cells producing EPO increases. In advanced CKD, the kidneys undergo end-stage fibrosis, during which these fibroblasts may transdifferentiate into myofibroblasts and lose their ability to produce appropriate amounts of EPO in response to hypoxia.26,27 However, these or other renal cells can be activated to increase their EPO output by the administration of therapeutic prolyl-hydroxylase inhibitors28 (Chap. 32), as indicated by the lower stimulated EPO production by anephric patients compared to those with end-stage renal disease and retained kidneys. Studies in animal models indicate that the impairment of EPO production in end-stage kidneys may be reversible and could be therapeutically restored.26,27
Inflammation is also a strong contributor to the pathogenesis of anemia of CKD. Patients who had renal disease with inflammation, as measured by increased serum CRP greater than 20 mg/L, required on the average 80 percent higher doses of EPO than patients with simple primary EPO deficiency from renal disease.29 In another study, patients with CRP greater than 50 mg/L reached lower concentrations of Hgb than patients with CRP less than 50 mg/L, despite higher doses of erythropoiesis-stimulating agents.30 Inflammation thus induces a state of relative resistance to EPO, contributing to the pathogenesis of anemia of CKD.
Hypoferremia, one of the defining features of AI, develops within hours of the onset of inflammation.1 Although previous studies of cytokine mediators of hypoferremia of inflammation were inconclusive, subsequent work31 indicates that the response is dependent on IL-6 which induces the iron-regulatory hormone, hepcidin.32 Unlike wild-type mice, mice deficient in either hepcidin33 or IL-634 do not become hypoferremic during turpentine-induced inflammation. In human hepatocyte cell cultures, IL-6 is a potent and direct inducer of hepcidin and neither IL-1 nor TNF-α share this activity. The central role of IL-6 is further indicated by the observation that IL-6-deficient mice do not acutely induce hepcidin in response to turpentine inflammation. Infusion of IL-6 into human volunteers induces hepcidin release within hours and causes concomitant hypoferremia.35 The IL-6–hepcidin axis now appears to be responsible for the induction of hypoferremia during inflammation. However, these studies do not exclude the potential contribution of other cytokines, including activin B and interferon-γ,13,36 to AI in human diseases or more complex mouse models. In support of multiple pathways of AI in a mouse model of inflammation, either the ablation of hepcidin or the ablation of IL-6 ameliorated the anemia, but neither restored normal Hgb concentration.37,38
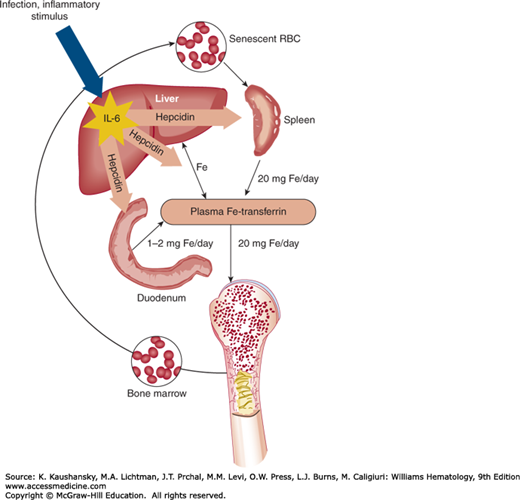
In the steady state, almost all of the approximately 20 to 25 mg of iron that daily enters the plasma iron/transferrin pool comes from macrophage recycling of senescent erythrocytes and from hepatocyte iron stores; only approximately 1 to 2 mg come from dietary iron. Only approximately 2 to 4 mg of iron is bound to transferrin but the entire daily iron flow transits through this compartment; thus, the iron in this pool turns over every few hours. During inflammation the release of iron from macrophages and probably also from liver stores is markedly inhibited.39,40,41,42,43,44,45 Studies in transgenic mice lacking hepcidin and mice overexpressing hepcidin indicate that the peptide is a negative regulator of iron release from macrophages and of intestinal iron uptake.46,47 During inflammation, IL-6 induces hepcidin production, which in turn inhibits iron release from macrophages (and probably from hepatocytes), leading to hypoferremia (Fig. 37–2). Hepcidin acts by binding to cell membrane-associated ferroportin molecules that are the only conduits for iron export, and inducing ferroportin internalization and degradation.48 As hepcidin concentrations increase, less and less ferroportin is available for iron export and the iron release into plasma from macrophages, hepatocytes, and enterocytes decreases.
As an intermediate step during the synthesis of heme, iron becomes incorporated into protoporphyrin IX. Zinc is an alternative protoporphyrin ligand. In iron deficiency, increased amounts of zinc are incorporated into protoporphyrin. In AI, zinc protoporphyrin is also increased.49 Insufficient iron reaches the sites of heme synthesis in developing erythrocytes, leading to the substitution of zinc. Moreover, the number of sideroblasts, nucleated erythrocyte precursors that stain for iron with Prussian blue, is decreased in AI.1 A further indication of the limiting role of iron in patients with AI but no evidence of iron deficiency is that coadministration of parenteral iron can resolve the resistance of AI to EPO.50,51 Attempts to treat AI with iron alone generally have been less successful, as iron became rapidly trapped in the macrophage compartment.1,52,53
In the context of anemia of CKD, increased zinc protoporphyrin and decreased reticulocyte Hgb is also characteristic of functional iron deficiency during intense bursts of erythropoiesis stimulated by pharmacologic doses of EPO derivatives.54
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


