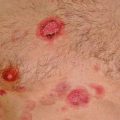
Fig. 6.1
(a and b) ALCL, ALK- with large tumor cells containing pleomorphic nuclei
6.3 Immunophenotype
The tumor cells of ALCL, ALK-positive and ALK-negative, universally express CD30 and a majority express CD43. Most ALK+ ALCL additionally express epithelial membrane antigen (EMA). Although ALCLs often express one or more general T-cell and cytotoxic T-cell antigens (i.e., CD2, CD5, CD4, TIA1, granzyme B), most have lost at least a proportion of the normal complement of antigens expressed by normal, mature T-cells (Benharroch et al. 1998; Foss et al. 1996). Most notably, CD3 is absent in up to 70 % of tumors (Kinney et al. 1993). Occasional cases have lost all detectable T-cell markers and express only CD30. Cases with a “null-cell” phenotype demonstrate genetic evidence of the T-cell lineage. Cases of ALK+ ALCL are distinguished by the expression of the kinase domain of the ALK oncoprotein which is detected with specific antibodies either in the cytoplasm only or both in the nucleus and cytoplasm (Pileri et al. 1997).
6.4 Genetics
Molecular studies demonstrate a clonal rearrangement of one or more genes encoding the T-cell receptor (TCR) in the tumor cells of ALCL regardless of the overall immunophenotype (Foss et al. 1996). ALK+ ALCL is additionally characterized by balanced translocations involving the 3′ portion of the ALK gene, which encodes the kinase domain of ALK, with the 5′ portion of a variety of partner genes, most commonly the NPM gene which encodes nucleophosmin (NPM) (Morris et al. 1995). The NPM-ALK fusion, as a result of the t(2;5)(p23;q35) (Gascoyne et al. 1999; Benharroch et al. 1998; Kadin and Morris 1998; Sandlund et al. 1994) balanced translocation, encodes a fusion protein with kinase activity and which promotes lymphoid transformation (Chiarle et al. 2003). ALK in these cases is usually expressed both in the cytoplasm and nucleus. In addition to NPM, there are at least eight additional known fusion partners with ALK− all of which preserve the ALK kinase domain and the resulting fusion proteins are usually expressed only in the cytoplasm. In contrast to ALK+ ALCL, ALK− ALCL is not distinguished by a unique chromosomal or known molecular abnormality.
6.5 Differential Diagnosis
The spectrum of morphologic appearances and immunophenotypes in ALCL frequently raise the differential diagnoses of classical Hodgkin lymphoma (cHL) and peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS). With the advent of better diagnostic markers of Reed-Sternberg cells that illuminate their B-lineage origin (such as PAX5), distinguishing ALCL from cHL has become more straightforward. In contrast, distinguishing ALK-negative ALCL from PTCL, NOS with CD30 expression has remained challenging despite recent attempts to clarify the distinction. The histologic appearance of the tumors cells and degree of expression of CD30 on the tumor cells remains a primary means of distinguishing these tumor types (Went et al. 2006). ALCL, as described here, is a systemic disease that may involve or present in the skin. However, care must be taken to distinguish cutaneous involvement by ALK-negative ALCL from primary cutaneous CD30+ T-cell lymphoproliferative disorders including primary cutaneous ALCL, lymphomatoid papulosis, and transformed mycosis fungoides.
6.6 Clinical Presentation
ALCL is an aggressive lymphoma which frequently presents in advanced clinical stage (III–IV) with systemic symptoms and extranodal involvement, as other PTCLs do (Falini et al. 1999; Stein et al. 2000). Bone marrow involvement is detected in up to 30 % of cases, being a relevant prognostic feature (Fraga et al. 1995; Mussolin et al. 2005; Kalinova et al. 2008).
Importantly, ALCLs display quite different clinical features depending on the expression of the ALK protein (Savage et al. 2008; Falini et al. 1999; Stein et al. 2000). In particular, ALK+ tumors most frequently occur among patients in the first or second decade of life, while ALK− ones are usually recorded among people aged 50–70 (Savage et al. 2008; Falini et al. 1999; Stein et al. 2000). Moreover, advanced-stage disease and B symptoms are slightly more common in ALK+ ALCL (Savage et al. 2008; Stein et al. 2000). B symptoms were observed in both groups, and patients with ALCL, ALK+ had significantly better performance status, and fewer had above normal LDH levels. Patients in both groups showed a nodal presentation, and >40 % of children with disseminated disease had inguinal lymph node involvement. Mediastinal involvement was less common than in Hodgkin lymphoma. Skin, bone, and soft tissues were commonly affected extranodal sites. Pelvic muscle involvement is not infrequent and can be mistaken for a soft tissue sarcoma. Central nervous system involvement seems rare in adults and especially in children with ALK+ ALCL. Bone marrow involvement is considered to be an uncommon event and can be difficult to detect on routine histologic examinations alone (Falini et al. 1999). ALCL, ALK+ is rare in patients after transplant (Costes-Martineau et al. 2002) and those infected with human immunodeficiency virus (Tirelli et al. 1995; Gabarre et al. 2001). In this setting, most ALCL cases appear to be related to the anaplastic variant of diffuse large B-cell lymphoma (Tirelli et al. 1995). Secondary ALCL may arise in the progression of other lymphomas, most commonly during the course of mycosis fungoides, PTCLs, Hodgkin lymphoma, or lymphomatoid papulosis (Stein et al. 2000), and is usually characterized by a poor prognosis (Salhany et al. 1988).
6.7 Prognostic and Predictive Factors
Comparison of ALK+ vs. ALK− ALCL in adult patients identified risk groups described in the International Prognostic Index (IPI) with a significant difference in favor of patients with ALK+, although those with an IPI score of three were in the poor risk regardless of ALK status (Savage et al. 2008). The T-cell prognostic index for T-cell (PIT) (Gallamini et al. 2004) was predictive of overall survival and failure-free survival in both groups. These clinical findings further support the inclusion of ALCL, ALK+ as a distinct entity in the new WHO classification. Interestingly, comparison of ALK+ and ALK− ALCL in patients >40 years of age revealed no difference in survival (Savage et al. 2008), suggesting that age is a predominant factor driving outcome difference. Suzuki et al. (2000) found CD56 expression to be a prognostic factor independent of IPI and ALK expression in multivariate analysis. In fact, in both ALK+ and ALK− subgroups, CD56+ cases showed a poorer prognosis than did CD56- cases.
Several series of childhood and adult ALCL have been reported but were often difficult to compare because of problems in defining entities, the heterogeneity of the treatments, or the lack of a common staging system (Brugieres et al. 1998; Dearden et al. 2011). St. Jude’s Hospital’s classification has been used only for children, whereas the Ann Arbor staging system and the IPI scoring are used for adults. Although ALK absence or presence in ALCL is useful, in association with the IPI score, for discriminating a patient’s prognosis and to evaluate the impact of treatment in adult patients, it is not applicable for children because 90 % of ALCLs at this age are ALK+ (Brugieres et al. 1998).
6.8 Treatment
6.8.1 Frontline Treatment
Multi-agent chemotherapy is the standard frontline treatment for patients with systemic ALCL, plus the addition of involved-field radiation therapy for patients who present with stage I–II locoregional disease (Dearden et al. 2011; Kwong et al. 2009; National Comprehensive Cancer Network (NCCN) 2012). However, the optimal chemotherapy regimen remains a bit unclear, mostly because of the lack of robust clinical trial data focusing on patients with ALCL and the need to extrapolate from trials of patients with PTCL.
A meta-analysis of studies examining anthracycline-based therapy in patients with PTCL showed good outcomes with this approach, similar to that seen in aggressive B-cell lymphomas (Abouyabis et al. 2011), and CHOP-based regimens have been shown to yield good responses and prolonged clinical benefit in patients with favorable prognosis ALC (Savage et al. 2008). However, data from the International Peripheral T-Cell Lymphoma Project show that outcomes in patients with ALCL are similar regardless of whether an anthracycline-based therapy is used as frontline chemotherapy (Vose et al. 2008). One possible approach to selecting an optimal treatment is to look more specifically at subgroup populations. The International Peripheral T-Cell Lymphoma Project found that, for patients with ALK+ disease, a CHOP-based regimen was shown to yield a 5-year overall survival rate of 75 % for patients with an IPI 0-1, but the rate fell to 25 % for patients with IPI ≥ 2. In patients with ALK− disease, higher-risk IPI also predicted a poorer outcome from CHOP-based chemotherapy: ALK− patients with IPI 0-1 showed a 5-year survival rate of 50 % vs. 18 % for patients with IPI ≥ 2 (Savage et al. 2008). Similar findings were seen in an examination of 363 patients with T-cell lymphoma enrolled on 7 prospective trials of different CHOP-based regimens from the German High-Grade Non-Hodgkin’s Lymphoma Study Group. Within this cohort, 78 patients (24 %) had ALK+ ALCL and 113 (35 %) had ALK− ALCL (Schmitz et al. 2010). Across all trials, the 3-year event-free and overall survival rates were 76 and 90 %, respectively, for patients with ALK+ ALCL, vs. 46 and 62 %, respectively, for patients with ALK− ALCL. In younger, favorable-risk patients, event-free survival rates were improved for patients treated with CHOP plus etoposide (CHOEP), while CHOP alone yielded better results for older patients with less favorable risk (Schmitz et al. 2010).
Of note, attempts at intensification by shortening the treatment interval in older patients or by escalating the treatment doses in younger patients did not improve outcomes (Schmitz et al. 2010). By contrast, the use of the intense NHL-Berlin-Frankfurt-Münster (BFM)-90 regimen, which had previously showed good results in a pediatric ALCL population (Seidemann et al. 2001), also showed good results in an adult ALCL population with a median age of 26 (range, 17–65). Based on these data, it would be reasonable to conclude that CHOP-21 is an appropriate frontline treatment strategy for older patients and those with less favorable ALCL (e.g., ALK− disease, higher-risk IPI), and CHOEP-14, CHOEP-21, or even the dose-intense BFM-90 protocol is an appropriate frontline treatment strategy for younger patients and those with more favorable ALCL (e.g., ALK+, lower-risk IPI).
In an attempt to improve outcomes beyond CHOP and dose intensification, researchers have explored alternative treatment strategies in select populations. For example, in 653 older patients with poorer-risk aggressive lymphoma, 22 of whom had ALCL, the French GELA compared standard CHOP with the ACVBP regimen (Tilly et al. 2003). Five-year event-free and overall survival rates were higher with ACVBP than with CHOP, even after adjusting for IPI and after excluding disease subtypes thought to have better outcomes. Rates of hematologic toxicity, infection, and the need for growth factor support were higher with ACVBP, again underscoring the importance of careful patient selection in determining the optional treatment approach (Tilly et al. 2003).
6.8.2 Consolidation with Autologous Stem Cell Transplant
Response rates in patients with ALK+ ALCL are typically high, and consolidation with autologous stem cell transplant (ASCT) is not recommended (Dearden et al. 2011; Kwong et al. 2009). By contrast, ASCT consolidation in patients with ALK− ALCL may be beneficial in some subpopulations. Although patients with ALCL, even those with ALK− disease, tend to fare better from transplantation than do patients with other types of PTCL (Jagasia et al. 2004), most trials enrol only a handful of patients with ALCL and fewer still differentiate between good prognosis ALK+ and poor-prognosis ALK− disease, making it difficult to truly assess the potential benefits of ASCT in these patients. Nevertheless, results of a few studies that specifically isolated patients with ALK− ALCL suggest that appropriately selected patients may benefit from this more aggressive treatment approach.
Extrapolating from a larger clinical trial, researchers with the Nordic Lymphoma Group evaluated 31 patients with ALK− ALCL who were treated with CHOP-14 (age 60–65) or CHOEP-14 (under age 60) followed by high-dose chemotherapy and ASCT. The response rate in transplanted patients was 96 % vs. 74 % for the group as a whole, and the 3-year overall survival rate was 73 % (Relander et al. 2012). Data from a German multicenter, prospective trial also showed good results with ASCT consolidation. In this trial, 83 patients with PTCL, 13 of whom had ALK− ALCL, were treated with CHOP followed by total body irradiation, high-dose chemotherapy, and ASCT. The estimated 3-year overall survival rate was 71 % for patients who underwent transplantation vs. 48 % for those who did not, and subgroup analysis showed no significant differences in overall survival between ALK− ALCL and other types of PTCL (Reimer et al. 2009).
As with other types of lymphoma, not all patients with ALK− ALCL achieve complete or partial response after frontline chemotherapy, yet this remains a key prognostic factor in determining outcome from ASCT (Hosing and Champlin 2011). In the trial from the Nordic group, only 77 % underwent ASCT, while in the German study, only 66 % did; in most cases, patients were excluded from treatment because they showed a poor response to frontline chemotherapy (Relander et al. 2012; Reimer et al. 2009). Of note, single-institution retrospective data from MD Anderson Cancer Center show that the presence of refractory disease, as indicated by an inability to achieve first complete response to induction therapy, independently predicts for a worse outcome from ASCT regardless of ALK status (Beitinjaneh et al. 2011). Thus, available data suggest that, for patients who show good response to frontline chemotherapy and who remain good candidates for transplantation, consolidation with ASCT can further improve outcomes (Dearden et al. 2011; Kwong et al. 2009). Nevertheless, until better regimens that are more effective at inducing high response rates are available, the potential benefits of ASCT consolidation will not be fully realized.
6.8.3 Second-Line Therapy
The relatively small population of patients with progressive systemic ALCL precludes large-scale clinical trials from definitively determining whether second-line chemotherapies, either multi-agent or single-agent, might be beneficial. Extrapolations are typically made from trials in patients with PTCL that may or may not relate to patients with ALCL, or even from trials in patients with NHL, in which patients with ALCL may represent only a tiny fraction of enrolees. Indeed, current clinical practice guidelines from the United States and Europe both describe the use of platinum-based multi-agent regimens, even though the trials evaluating those regimens were often primarily done in patients with aggressive B-cell NHL (Dearden et al. 2011; Kwong et al. 2009). Thus, although these and other chemotherapy regimens can be considered in patients with systemic ALK− ALCL, the outcomes remain unknown and their potential benefit remains difficult to assess.
The benefit of salvage SCT is also unclear. A review of records on patients with T-cell lymphoma by the Center for International Blood and Marrow Transplant Research suggest that a greater number of prior chemotherapy regimens and chemoresistance independently predict for worse outcomes from both autologous and allogeneic SCT, indicating that these treatment modalities should be considered earlier in the course of disease (Schmitz et al. 2010). Nevertheless, studies in patients with ALCL seem to suggest that certain patient populations can benefit from autologous and allogeneic SCT in the salvage setting. Trials with salvage ASCT in patients with ALK− ALCL have shown little benefit for its use in this population. In a series of 16 patients with ALK− ALCL, only 1 patient showed long-term disease-free survival, while 14 showed relapsed after a median of only 12 weeks despite achieving complete or partial response after salvage chemotherapy (Zamkoff et al. 2004). By contrast, patients with ALK+ disease seem to fare well from salvage ASCT: in one series, patients with ALK+ ALCL showed very high event-free survival rates (Jagasia et al. 2004). Thus, in patients with ALK+ disease, salvage ASCT can prolong remission but the treatment modality seems to offer little benefit to patients with ALK− ALCL.
Salvage allogeneic SCT might be an option for patients with systemic ALK− ALCL. In particular, patients with ALK− ALCL who underwent allogeneic SCT with reduced-intensity conditioning showed good response. In a series of 17 patients with PTCL, after at least 10 months’ follow-up, 1 of 4 patients with ALK− ALCL had stable disease, 1 achieved partial response after donor lymphocyte infusion, and 2 achieved complete response (Corradini et al. 2004). Of note, the partial responder and one of the complete responders had relapsed after a prior autologous SCT, underscoring that patients with chemosensitive disease can continue to benefit from further treatment modalities.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


