Malaria is an acute, chronic, or recurrent febrile disease caused in humans by four species of
Plasmodia: Plasmodia vivax, Plasmodia falciparum, Plasmodia malariae, and
Plasmodia ovale. Infections with
P. falciparum are the major form of malaria in Africa and Southeast Asia, whereas
P. vivax is most common in Central America and India. These protozoan microorganisms are capable of parasitizing erythrocytes and other body tissues. Malaria is spread by female mosquitoes of the genus
Anopheles (
Fig. 32.1). The sexual phase of the
Plasmodium life cycle takes place within the mosquito. The semitropical and tropical endemic distribution of malaria corresponds to the distribution of the vector.
On a worldwide basis, malaria is the most prevalent of all serious diseases; it has been estimated that approximately 2.5 billion people are at risk for malaria, and that approximately 500 million people are infected with
P. falciparum.
1,
2 Ninety percent of the deaths is in African children.
3 Malaria has not been endemic in the United States since the 1940s, but approximately 1,000 cases have been reported each year since 1985, and there has been a steady increase in the number of cases reported annually.
4 Malaria can also be transmitted by blood transfusions
5 or by sharing needles among intravenous drug abusers.
6,
7
Clinical Manifestations
After the initial exposure to malaria, some patients are completely asymptomatic, whereas others have nonspecific flulike symptoms that mimic a viral illness.
8 Classically, the most prominent clinical manifestations are recurrent paroxysms of chills and fever with temperatures as high as 105° to 106°F (40.5° to 41°C) associated with malaise, headache, vomiting, and other systemic symptoms. The paroxysms tend to recur regularly every 36 to 72 hours. They are most prominent with
P. malariae infections and much less with
P. falciparum. Splenomegaly is noted in about one half of patients during early stages of disease
8 and becomes more common later. Jaundice and hepatomegaly may develop in later stages of the illness.
Of the various malaria species,
P. falciparum infection causes the most morbidity and mortality. In the acute stage, it can be associated with increasing parasitemia, hypotension, malignant hyperthermia, and death. In addition,
P. falciparum malaria is associated with cerebral, pulmonary, and renal complications.
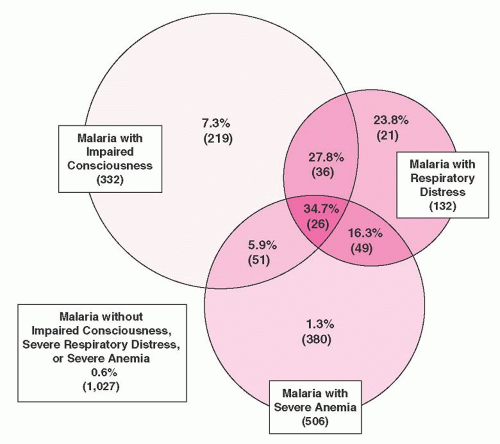
9 The overall mortality from a study of over 1,800 children with malaria in Kenya was 3.5%; in 84% of cases death occurred within 24 hours of admission.
10 The most important prognostic factors for death were impaired consciousness and respiratory distress (
Fig. 32.2). Severe anemia alone did not affect prognosis.
3,
10Anemia is common in malaria.
11,
12,
13,
14 It is particularly characteristic of
P. falciparum malaria because of the greater extent of red cell parasitization with this species. With uncomplicated
P. falciparum malaria, moderately severe anemia is seen in approximately 20% of previously healthy patients during or after the first infection.
15 Complete eradication of malaria parasites from the blood may take months to years, particularly in areas of high transmission; immunity to malaria is slowly acquired. In tropical areas, anemia tends to be most prevalent and most severe in children from 1 to 5 years of age,
16 whereas only moderate anemia is usually noted in adolescents and adults.
In children, the circulating parasite count is inversely proportional to the hematocrit.
17 Leukocyte numbers may be normal, but patients often have leukopenia. Thrombocytopenia has been observed in about two thirds of patients with
P. falciparum malaria,
9,
16 often associated with splenomegaly.
18The most serious hematologic complication of malaria is acute intravascular hemolytic anemia (blackwater fever), which occurs as a rare event in the course of infection by P. falciparum. The clinical manifestations are fulminating, the intravascular hemolysis being associated with prostration, vomiting, chills, and fever. Hemoglobinemia, hemoglobinuria, and hyperbilirubinemia are consistent features, and in the most severe episodes, acute oliguric renal failure supervenes.
Pathogenesis
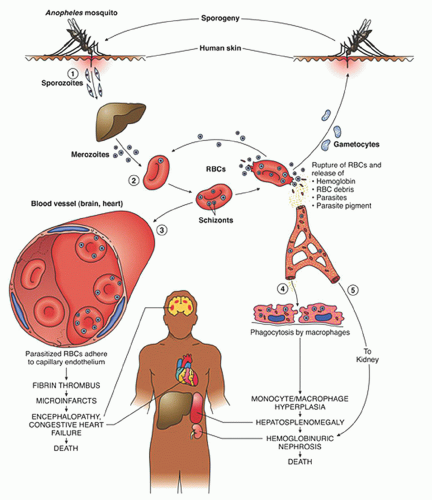
After a bite from the female
Anopheles mosquito, sporozoites introduced into the circulation go to the liver parenchyma where they proliferate into thousands of merozoites (
Fig. 32.1). The duration of this liver development stage varies between species. The infected hepatocytes next release merozoites into the bloodstream where they invade erythrocytes.
The ability of various
Plasmodia to infect red cells is related to their attachment to specific membrane receptors. Of the species that infect humans,
P. vivax and
P. ovale invade only reticulocytes.
P. malariae invades mature red cells, and
P. falciparum invades erythrocytes of all ages. As a result, the proportion of cells parasitized in
P. vivax malaria rarely exceeds 1%, whereas as many as 50% of cells may be affected in
P. falciparum malaria. It is of interest that
P. vivax invades only Duffy blood-group-positive red cells
19; in West Africa where the Duffy antigen is missing on red cells,
P. vivax malaria is nonexistent.
P. falciparum apparently has two receptors: one that binds to sialic acid groups on the erythrocyte membrane protein glycophorin and another that
binds to a trypsin-sensitive, nonsialated ligand.
20,
21,
22 Other proposed parasite receptors include surface heparinlike molecules
23 and complement receptor type-1.
24Further development of the parasite within red cells is along one of two pathways, either asexual or sexual differentiation (
Fig. 32.1). Sexual forms, or gametocytes, continue their development within mosquitoes. The asexual differentiation of parasites in red cells proceeds from young ring forms through trophozoites to produce schizonts containing 6 to 32 merozoites (
Fig. 32.3). In the process, parasites use 25% to 75% of the hemoglobin of the cell.
25 The intraerythrocytic phase lasts 24 to 72 hours, depending on the species. The schizonts then lyse, the cell ruptures, and the merozoites are released to invade other cells, thereby continuing the erythrocyte cycle. The simultaneous rupture of billions of schizonts from red cells is associated with the classic paroxysms of malarial fever.
Erythrocytes parasitized by certain strains of
P. falciparum develop electron-dense knobs that mediate the attachment of the infected red cells to venules.
26,
27,
28 Such sequestration of parasiteinfected RBC creates an obstruction to tissue perfusion. In addition, the sequestration in venules prevents parasitized cells from entering the splenic circulation, thereby evading destruction and enhancing merozoite development; this phenomenon may be a factor in the rapid development of anemia in severe infections.
29The anemia in malaria is due to a combination of factors that include parasite-mediated RBC destruction, splenic removal of
infected RBCs, and decreased red cell production (
Table 32.1). Hemoglobin digestion and cell disruption by the parasite are clearly the major causes of hemolysis.
11,
12,
30 The malarial pigment hemozocin, a product of hemoglobin degradation,
25 also inhibits erythropoiesis.
31,
32The role of the spleen in red cell destruction is related to the decreased deformability of RBCs infected with
Plasmodia,
33,
34,
35 erythrocyte retention in the red pulp, prolonged exposure to splenic macrophages, and removal of parasitized cells or “pitting” of the parasite, with consequent damage to the cell.
36,
37,
38 A similar process of macrophage-mediated destruction of parasitized RBCs occurs in the marrow sinusoids.
39 Moreover, normal nonparasitized RBCs have a shorter survival in malaria, presumably a consequence of hypersplenism and hyperactive macrophages.
40Even after complete clearance of the parasites, hemolysis may persist for 4 to 5 weeks.
41 A complement-mediated process may be responsible in part, and the direct antiglobulin test is often positive.
42Dyserythropoiesis with characteristic morphologic findings also occurs in malaria. It is thought that this contributes to the slow recovery seen after a single malarial attack and also to the persistence of anemia in individuals with chronic parasitemia.
13,
43,
44Anemia often persists for weeks following treatment of malaria, and this results in part from relative marrow failure, as occurs in association with other forms of infection (see
Chapter 41).
12,
41 Abnormalities in several of the cytokine mediators of the anemia of chronic disorders (tumor necrosis factor, interferon gamma) have been reported in patients with severe
P. falciparum malaria.
45,
46,
47,
48,
49Serum erythropoietin levels often are inadequate for the degree of anemia.
50,
51,
52 The bone marrow response to erythropoietin also appears to be impaired.
53 Circulating concentration of the iron regulatory peptide hepcidin, a key mediator of the anemia of chronic disease, is elevated early in the course of malaria.
54,
55 This results in impaired iron mobilization.
56,
57The percentage of reticulocytes tends to be low during active infection and increases transiently after effective treatment. In
P. vivax malaria, however, the low reticulocyte count may be explained in part by the increased affinity of the organism for immature erythroid precursors and reticulocytes.
58 Infection by
P. vivax appears to be retricted to reticulocytes.
59The pathogenesis of acute intravascular hemolysis (blackwater fever) remains uncertain, and currently this complication is less commonly seen, although it still occurs.
60,
61 Blackwater fever does not reflect an unusual degree of parasitemia. In many historical cases, the acute intravascular hemolysis appears to have been precipitated by quinine ingestion
62,
63; quinine, for many years the primary treatment agent for malaria, may act as
a hapten, becoming antigenic after interacting with the red cell. However, cases involving untreated individuals also have been reported.
9 Some episodes thought to represent blackwater fever may have resulted from the use of primaquinelike drugs in G6PD-deficient people.
Certain inherited red cell disorders appear to confer resistance to malaria, either by inhibiting parasitic invasion or by slowing intracellular growth. It is thought that these phenomena may contribute to increased prevalence of such inherited diseases because of their effects on survival (balanced polymorphism). These disorders include sickle cell trait,
64,
65,
66 G6PD deficiency,
67,
68,
69,
70 thalassemia,
71,
72 hemoglobin E variants,
70 hemoglobin C variants,
73 ovalocytosis of the Melanesian (Malayan) type,
74 and lack of the Duffy blood group antigen.
19
 Clinical Flow Cytometry
Clinical Flow Cytometry



