INTRODUCTION
SUMMARY
Sideroblastic anemias are characterized by the presence of ring sideroblasts in the marrow. These cells are erythroid precursors that have accumulated abnormal amounts of mitochondrial iron. A variety of abnormalities of porphyrin metabolism in affected erythroid cells have been documented. Hereditary sideroblastic anemias are usually X linked, as the result of mutations in the erythroid form of 5-aminolevulinic acid synthase. Inherited autosomal and mitochondrial forms are seen, occasionally. Acquired sideroblastic anemias can occur as a result of the ingestion of drugs, alcohol, or toxins such as lead or zinc, or copper deficiency. Patients with acquired sideroblastic macrocytic anemia and variable degrees of thrombocytopenia and leukopenia from copper deficiency have been recognized more frequently; the hematologic abnormalities typically resolve after copper replacement. Ring sideroblasts are also a feature of myelodysplastic neoplasms, and are discussed in Chap. 87. Some patients with sideroblastic anemia may respond to pharmacologic doses of pyridoxine. Iron loading is common in the sideroblastic anemias and can be treated by phlebotomy when the anemia is mild or with iron chelators (Chap. 43) when it is more severe.
DEFINITION AND HISTORY
Sideroblastic anemias are a heterogeneous group of disorders that have as a common feature the presence of: (1) large numbers of pathologic sideroblasts in the marrow, which characteristically display abnormal mitochondrial iron accumulation is in a circumnuclear position in erythroblasts; these are referred to as ringed sideroblasts; (2) ineffective erythropoiesis; (3) increased levels of tissue iron; and (4) varying proportions of hypochromic erythrocytes in the blood. They may be acquired or hereditary (Table 59–1).1,2
Acquired monoclonal sideroblastic anemia is a neoplastic disease; that is, a clonal cytopenia or oligoblastic myelogenous leukemia that can progress to acute leukemia. This subject is considered in Chap. 87, Myeloid Neoplasms, in which ringed sideroblasts are a common phenotypic feature. Acquired polyclonal sideroblastic anemia may also develop as a result of the administration of certain drugs, exposure to toxins, or coincident to neoplastic or inflammatory disease. Hereditary sideroblastic anemias include X-linked, autosomal, and mitochondrial entities. Occasionally, a patient with familial disease develops a myelodysplastic syndrome later,1,2 but with these rare exceptions, the disorders are distinct and do not coexist or evolve one from the other.
Acronyms and Abbreviations:
ABCB7, ATP-binding cassette; ALA, 5-aminolevulinic acid; ALAS2, gene encoding ALA synthase 2; CPO, coproporphyrinogen oxidase; FECH, ferrochelatase; Fe-S, iron-sulfur; GLRX5, glutaredoxin 5; MLASA, mitochondrial myopathy and sideroblastic anemia; PUS1, pseudouridine synthase 1 gene; SLC25A38, mitochondrial carrier family gene; STEAP 3, six-transmembrane epithelial antigen of prostate 3-ferric reductase; TfR, transferrin receptor; tRNA, transfer RNA; XLSA/A, X-linked sideroblastic anemia, associated with ataxia.
Although the perinuclear distribution of siderotic granules in the erythroblasts of patients with various types of anemia was described in 1947,3,4 the concept of sideroblastic anemia as a generic designation was not generally accepted until the publications of Björkman,5 Dacie,6 Heilmeyer,7,8 Bernard,9 and Mollin.10 After these descriptions of the acquired, “primary adult form of refractory sideroblastic anemia,”5,6 similarity to the morphologic and erythrokinetic changes in hereditary (sex-linked) hypochromic anemia was recognized. Cooley11 described a patient with an anemia with ovalocytosis who was shortly thereafter shown to have inherited a hereditary sex-linked disorder12 that we now know resulted from the mutation of erythroid-specific aminolevulinic acid (ALA) synthase, ALAS2.13 Autosomally inherited cases were also described,14 and prominent sideroblastic changes of the marrow were found in Pearson marrow-pancreas syndrome (Chap. 36), a disorder that is caused by mutations of mitochondrial DNA.15,16,17,18,19 Sideroblastic anemia can also be associated with a wide variety of diseases,20 therapy with antituberculosis drugs,21,22 and lead intoxication.23,24,25,26 In some patients, the anemia responded to large doses of pyridoxine and was designated “pyridoxine-responsive anemia.”10,27,28,29 These “secondary” acquired disorders were then incorporated into the classification.
EPIDEMIOLOGY
All of the hereditary forms are rare, and no particular ethnic predilection is known. Drug-induced forms occur sporadically among subjects taking the drugs listed in Table 59–1.
ETIOLOGY AND PATHOGENESIS
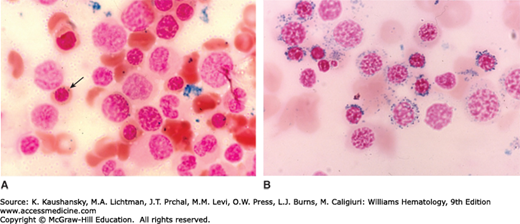
Sideroblasts are erythroblasts containing aggregates of non–heme iron that appear as one or more Prussian blue–positive granules on light microscopy.30 The morphology of these cells in normal and abnormal states is discussed in detail in Chap. 31. In normal marrow, virtually every erythroblast has siderosomes, iron-containing organelles that are demonstrable by transmission electron microscopy. Light microscopy of Prussian blue–stained marrow aspirates or biopsy sections is a relatively insensitive method to identify these structures. One can usually identify approximately 25 to 35 percent of erythroblasts with one to three very fine Prussian blue–stained granules in the cytoplasm of a well-prepared marrow sample. Pathologic sideroblasts may be of two types: erythroblasts with a marked increase in the number and size of siderotic granules in the cytoplasm, compared to normal erythroblasts, or ringed sidero-blasts. Ringed sideroblasts are the hallmark of the sideroblastic anemias. In contrast to the normal cytoplasmic location of siderotic granules, the pathologic sideroblasts in the sideroblastic anemias have large amounts of iron deposited as dust- or plaque-like ferruginous micelles between the cristae of mitochondria (Fig. 59–1).31 The iron-loaded mitochondria are distorted and swollen, their cristae are indistinct, and identification of mitochondria may itself be difficult. In humans, the mitochondria of the erythroblast are distributed perinuclearly,23 which accounts for the distinctive “ringed” sideroblast identified by Prussian blue staining when mitochondrial iron overload is present (Fig. 59–1). The morphologic features that characterize pathologic sideroblasts in various disorders have been summarized.32
Figure 59–1.
Marrow films. A. Normal marrow stained with Prussian blue. Note several erythroblasts without apparent siderotic (blue-stained) granules. The arrow indicates erythroblast with several very small cytoplasmic blue-stained granules. It is very difficult to see siderosomes in most erythroblasts in normal marrow because they are often below the resolution of the light microscope. B. Sideroblastic anemia. Note the florid increase in Prussian blue–staining granules in the erythroblasts, most with circumnuclear locations. These are classic examples of ringed sideroblast, which are by definition pathologic changes in the red cell precursors. In some cases, cytoplasmic iron granules are also increased in size and number, also a pathologic change. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
The pathogenesis of most of the sideroblastic anemias is not well understood.33,34 It is not clear whether the basic mechanism by which abnormal accumulations of intramitochondrial iron occurs is the same in inherited and acquired forms of the disease. However, it seems appropriate, given the present state of knowledge, to discuss both forms together. The pathogenesis of the disorder may be viewed from two standpoints: the underlying biochemical lesions and the mechanism(s) of the anemia itself.
In the search for the biochemical lesions responsible for the development of sideroblastic anemia, attention has been focused upon an intramitochondrial defect in heme synthesis and on possible disturbances in pyridoxine metabolism.
The role of defects in heme biosynthesis have occupied central stage since the early studies of Garby and colleagues,35 who postulated that such a defect might exist; they demonstrated that the level of free erythrocyte protoporphyrin was decreased. Subsequently, a variety of abnormalities of the levels of precursors and of their rate of incorporation into heme was documented (Chap. 58).36,37,38,39,40,41 However, the findings have not all been consistent, as levels of free erythrocyte protoporphyrin have often been increased,42,43 not diminished. The role of mitochondria in the etiology of sideroblastic anemia gained further credence when mutations of the mitochondrial genome were found in patients with Pearson syndrome.15,16,17,18,19
Shortly after the identification of erythroid-specific ALA synthase (ALAS2, the first enzyme in heme synthesis; Fig. 59–2), it became apparent that most patients with hereditary X-linked sideroblastic anemias (XLSAs) had mutations in the ALAS2 gene.44,45,46 However, a proportion of patients with congenital sideroblastic anemia had autosomal recessive inheritance. At least some such patients have a defect in the gene encoding the erythroid-specific mitochondrial carrier protein, SLC25A38.47 This transporter is important for the biosynthesis of heme in eukaryotes and it was proposed that this protein may be translocating glycine into mitochondria (Fig. 59–2).47 Hence, SLC25A38 defects would be expected to generate a phenotype identical to that seen in patients with defects in ALAS2. One can speculate that, in erythroid cells, a common control mechanism exists that regulates acquisition of the two substrates for heme synthesis (iron and glycine).
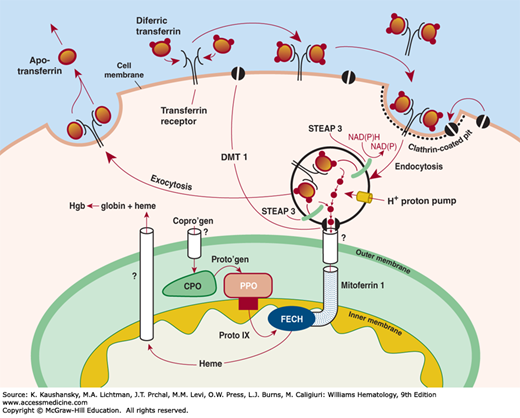
Figure 59–2.
Schematic of iron uptake from transferrin and its delivery to the hemoglobin (Hgb) molecule. Extracellular differic transferrin is bound by the membrane-bound transferrin receptor (TfR) and internalized via receptor-mediated endocytosis into an endosome. Iron is released from transferrin by a decrease in pH (~pH 5.5), reduced by STEAP 3 (six-transmembrane epithelial antigen of prostate 3-ferric reductase), following which the metal is transported through the endosomal membrane by DMT 1. In erythroid cells, more than 90 percent of iron must enter mitochondria wherein ferrochelatase (FECH), the enzyme that inserts Fe2+ into protoporphyrin IX (Proto IX), resides on the inner leaflet of the inner mitochondrial membrane. The transport of coproporphyrinogen (Copro’gen) into mitochondria is not fully understood. Neither mechanisms nor the regulation of the transport of heme from mitochondria to globin polypeptides are known; however, it has been proposed that a carrier protein, heme binding protein 1 (gene: HEBP1), is involved in this process. CPO, coproporphyrinogen oxidase; NAD(P), nicotinamide adenine dinucleotide phosphate; NAD(P)H reduced form of nicotinamide adenine dinucleotide phosphate; PPO, protoporphyrinogen oxidase. (Reproduced with permission from Anderson GJ, McLaren G: Iron Physiology and Pathophysiology in Humans, New York, NY: Humana Press; 2012.)
Hereditary sideroblastic anemia with spinocerebellar degeneration with ataxia is a rare X-linked syndrome that appears to be distinct from the other forms of sideroblastic anemia.48,49,50,51 It is caused by mutation of ATP-binding cassette (ABCB7).48,52,53
Heteroplasmic point mutations in subunit 1 of the mitochondrial cytochrome oxidase have been documented in some patients with sideroblastic anemia.54,55,56
Rare autosomal forms of inherited sideroblastic anemia have also been reported,57,58 including those with a deficiency of uroporphyrinogen decarboxylase59,60 and ferrochelatase (FECH)36,41,61,62,63 enzymes, both necessary for the synthesis of heme (Chap. 58). The other reported defects of ferrochelatase could result from the inhibitory effect of mitochondrial iron overload on enzyme activity.41 A defect in coproporphyrinogen oxidase (CPO) could not be confirmed by direct measurement.64
An unusual phenotype with of inherited sideroblastic anemia, developmental delay with variable neurologic defects and B-cell lymphopenia with hypogammaglobulinemia was reported of yet unknown etiology.65
A role for pyridoxine has been supported by the demonstration that pyridoxine deficiency in animals is a prototype of sideroblastic anemia.31 Sideroblastic anemia can be induced by drugs that reduce the level of pyridoxal phosphate in blood, which decreases the ALAS2 activity in normoblasts.22,36,40 Moreover, certain sideroblastic disorders, although not a result of pyridoxine deficiency, are nonetheless responsive to pharmacologic doses of pyridoxine.44,66,67,68 Pyridoxal phosphate is a necessary coenzyme for the initial reaction of protoporphyrin synthesis, the condensation of glycine and succinyl coenzyme A to form ALA, a reaction mediated by ALA synthetase (Chap. 58). Furthermore, pyridoxal phosphate is a factor in the enzymatic conversion of serine to glycine (Chap. 41). This reaction generates a form of folate coenzyme necessary for the formation of thymidylate, an important step in DNA synthesis. Pyridoxal 5′-phosphate, the active form of the coenzyme, must itself be enzymatically synthesized from pyridoxine. Deficiencies in its biosynthesis have also been invoked as the possible cause of certain sideroblastic anemias,27,69 but direct measurements of pyridoxal kinase failed to confirm that the postulated lesion was present.70
Other Metabolic Defects and Acquired Associations with Sideroblastic Anemia of Uncertain Significance
Increased levels of uroporphyrinogen 1 synthase are commonly encountered in patients with sideroblastic anemias.39 Alcohol, a common cause of secondary sideroblastic anemia, inhibits heme synthesis at several steps.38 Dramatically altered activity ratios of a wide diversity of enzymes have been described,71,72 for example, elevated arginase activity.
Sideroblastic anemia has been found in a patient with apparent antibody-mediated red cell aplasia.73 There are alterations in red cell antigen patterns frequently with an increase of i and a loss of A1 antigens (Chap. 136).74 Similar findings occur in certain hereditary and acquired refractory anemias with cellular marrows but without ringed sideroblasts.72 Such dyscrasias are also characterized by ineffective erythropoiesis and, except for the lack of ringed sideroblasts, may in some instances be virtually indistinguishable from their sideroblastic counterparts.75
Iron accumulation within mitochondria is an unusual pathologic phenomenon occurring only in erythroblasts of patients with sideroblastic anemias and, to a much lesser degree, in cardiomyocytes of patients with Friedreich ataxia.76,77 Mitochondrial iron accumulation has not been demonstrated in patients with either primary or secondary iron overload. The pathophysiology of ring sideroblast formation in patients with ALAS2 defects and those resulting from inhibitors of porphyrin biosynthesis (see Table 59–1) is likely because of the unique aspects of the regulation of iron metabolism and heme synthesis in erythroid cells (Chap. 58).78 These differences can account for the accumulation of non–heme iron in erythroid mitochondria of sideroblastic anemia patients. In hemoglobin-synthesizing cells, iron is specifically targeted toward mitochondria that avidly take up iron even when the synthesis of protoporphyrin IX is suppressed (Chap. 58).79,80,81,82 In contrast, nonerythroid cells store iron in excess of metabolic needs within ferritin.83 Hence, erythroid-specific mechanisms and controls are involved in the transport of iron into mitochondria in erythroid cells, but the nature of these processes, including the role of mitoferrin 1 (Chap. 42); an inner mitochondrial membrane protein, which presumably provides Fe2+ to FECH,84 is poorly understood. The transferrin-bound iron is used for hemoglobin synthesis78,82 with a high degree of efficiency and is targeted into erythroid mitochondria (Chap. 52), and because no intermediate for cytoplasmic iron transport has ever been identified in erythroid cells, the following hypothesis of intracellular iron trafficking in developing red cells has been proposed (see Fig. 59-2 from Chap. 52). This model postulates that iron released from transferrin in the endosome is passed directly from protein to protein until it reaches FECH, which incorporates Fe2+ into protoporphyrin IX85 in the mitochondrion. Such a transfer bypasses the cytosol, as the movement of iron between proteins could be mediated by a direct interaction of the endosome with the mitochondrion.78,86 The results of supporting experiments revealed that (1) iron, delivered to mitochondria via the transferrin–transferrin receptor (TfR) pathway, is unavailable to cytoplasmic chelators87,88
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


