INTRODUCTION
SUMMARY
T cells represent an important component of the host response to pathogens and tumors. Adoptive T-cell therapy, in which T cells are isolated or engineered to be specific for molecules expressed on diseased cells and administered to patients, has shown efficacy in infections and malignancy. Clinical applications of T-cell therapy have been facilitated by identification of target antigens expressed by viruses and tumors, improvement in strategies for the isolation and genetic engineering of antigen-specific T cells with intrinsic qualities that enable their persistence in vivo, and recognition that transferring T cells into a lymphopenic environment improves the efficiency of cell transfer and treatment efficacy. Insights into the obstacles to routinely achieving an effective antitumor response either by T-cell therapy or vaccination have been derived from careful analysis of clinical trials, and further development of immune cell therapy combined with interventions that target specific regulatory or inhibitory pathways that are present in tumor microenvironments and impede effective immunity represent promising areas for future applications.
Acronyms and Abbreviations:
ALL, acute lymphatic leukemia; BCMA, B-cell maturation antigen; CAR, chimeric antigen receptor; cDNA, complementary DNA; CDR3, complementarity determining region 3; CEA, carcinoembryonic antigen; CLL, chronic lymphatic leukemia; CMV, cytomegalovirus; CRS, cytokine release syndrome; CTL, cytotoxic T lymphocyte; DLI, donor lymphocyte infusion; E, early viral protein; EBV, Epstein-Barr virus; EGFR, epidermal growth factor receptor; ERBB2IP, erbb2 interacting protein; GD2, disialoganglioside; GVHD, graft-versus-host disease; GVL, graft-versus-leukemia; HHV-6, human herpes virus-6; HLA, human leukocyte antigen; HSCT, hematopoietic stem cell transplantation; HSV, herpes simplex virus; HSV-TK, herpes simplex virus thymidine kinase; iCasp9, inducible caspase-9; IE, immediate early viral protein; IFN-γ, interferon-γ; IL, interleukin; L1CAM, L1-cell adhesion molecule; LCL, lymphoblastoid cell line; LPD, lymphoproliferative disease; mAbs, monoclonal antibodies; mHAgs, minor histocompatibility antigens; MHC, major histocompatibility complex; PBMC, peripheral blood mononuclear cell; PD-1 receptor, programmed death-1 receptor; PML-RARα, promyelocytic leukemia–retinoic acid receptor α protein; scFV, single-chain variable fragment; SNP, single nucleotide polymorphism; TCM, central memory T cell; TCR, T-cell receptor; TE, effector T cell; TEM, effector memory T cell; Th, T helper; TIL, tumor-infiltrating lymphocyte; TREG, regulatory T cell; TSCM, T memory stem cell; WT-1, Wilms tumor antigen-1.
ADOPTIVE CELLULAR THERAPY OF VIRAL DISEASES
Two broad subsets of antigen-specific T cells cooperate to terminate acute viral infections and control reactivation of latent viruses. CD8+ cytotoxic T lymphocytes (CTLs) recognize viral peptides presented by major histocompatibility complex (MHC) class I molecules and lyse infected cells, and produce inflammatory cytokines. CD4+ T-helper (Th) cells recognize viral peptides presented by class II MHC molecules and produce cytokines that amplify T-cell responses and promote B-cell proliferation and antibody production. A deficiency of CD8+ and CD4+ Th cells occurs after allogeneic hematopoietic stem cell transplantation (HSCT) as a consequence of the administration of intensive chemoradiotherapy, anti–T-cell monoclonal antibodies (mAbs), and/or immunosuppressive drugs, and these patients are at risk for life-threatening viral infections.1,2,3,4,5 Clinical trials have shown that adoptive T-cell therapy has antiviral activity against cytomegalovirus (CMV), Epstein-Barr virus (EBV), and adenovirus infection in immunocompromised allogeneic HSCT recipients.6,7
The first application of adoptive T-cell therapy with antigen-specific T cells in humans was to treat CMV infection after allogeneic HSCT. CMV is a DNA virus that infects hematopoietic progenitors, monocytes, and endothelial cells.8 To evade complete elimination by host immunity, CMV encodes proteins that interfere with antigen presentation in cells that contain replicating virus, and establishes latency.9 Normal CMV+ individuals maintain high levels of CD8+ and CD4+ T cells that are specific for CMV antigens, and these responses are essential to control infection.10,11,12 CMV frequently reactivates in individuals with a T-cell immunodeficiency, such as after allogeneic HSCT and solid-organ transplantation, and contributes to morbidity and mortality. Lymphocytopenia and a deficiency of functional CMV-specific T cells persist for several months in many HSCT recipients, including cord blood transplant recipients, and CMV reactivation is frequent.13,14,15 Antiviral drugs are used to suppress CMV,16,17,18 but often provide only temporary control of reactivation, and restoration of immune function is essential to contain CMV infection.13,19,20,21
Studies of the specificity of CMV-specific T cells isolated from immunocompetent CMV seropositive individuals identified antigens to target in T-cell therapy.15,21 A majority of CD8+ CTLs elicited by in vitro stimulation with autologous CMV-infected cells recognize virion proteins, including the pp65 and pp150 matrix proteins that are introduced into the cytoplasm of cells immediately following viral entry and processed and presented for T-cell recognition. Although virion proteins are important targets for CD8+ T cells, stimulation of peripheral blood mononuclear cells (PBMCs) from normal CMV+ donors with panels of CMV peptides, or with cells infected with a CMV strain in which the immune evasion genes had been deleted, identified significant CD8+ T-cell responses to intermediate-early (IE) or early (E) viral proteins.10,11 IE and E are not efficiently presented to T cells in vitro by cells replicating wild-type CMV, but evidence from animal models suggests that IE-specific T cells recognize cells that are reactivating CMV from latency.22 Thus, reconstitution of responses both to virion and IE or E antigens may be necessary to restore control of both the latent and replicating pools of virus in immune-deficient hosts.23
CD4+ Th cells are required for optimal CD8+ CTL responses and may eliminate CMV-infected cells that express class II MHC in vivo. Studies using recombinant CMV proteins or peptide panels have identified CD4+ T-cell responses to pp65, IE-1, glycoprotein B, and the major capsid protein (UL86) in normal CMV+ individuals.10,24,25
The application of T-cell therapy for CMV in allogeneic HSCT recipients required the development of approaches to reliably isolate CMV-specific T cells from the stem cell donor, and to remove potentially alloreactive T cells that could cause graft-versus-host disease (GVHD). The first clinical trial of T-cell therapy employed CD8+ CMV-specific T-cell clones that were isolated and expanded by in vitro culture of donor lymphocytes with autologous CMV-infected fibroblasts.26 The donor-derived T-cell clones were screened to exclude alloreactivity with noninfected recipient cells prior to adoptive transfer, to minimize the risk of causing serious GVHD. In a phase I study, 14 allogeneic HSCT recipients received four escalating weekly intravenous doses (3.3 × 107 to 1 × 109/m2) of CD8+ CMV-specific CTL clones as prophylaxis for CMV disease. The treatment did not cause toxicity or exacerbate GVHD, CMV-specific cytolytic activity was increased after therapy to levels equivalent to those in the donor, and transferred CTLs persisted for more than 12 weeks.27 None of the 14 patients developed CMV viremia or disease, which in the absence of antiviral drug therapy was expected to occur in approximately 50 percent or 40 percent of these patients, respectively.27
The results of the initial trials with CMV-specific T-cell clones suggested this approach can provide an alternative to antiviral drugs for controlling CMV infection after HSCT. However, the isolation and propagation of antigen-specific T-cell clones requires specific expertise and is time-consuming. Culture methods for enrichment of polyclonal CMV-specific T cells circumvent prolonged ex vivo manipulation and T-cell expansion and enabled broader application of this approach (Fig. 26–1).28,29 Clinical trials with polyclonal CD4+ and/or CD8+ T-cell lines generated by short-term culture confirmed the efficacy of adoptive therapy for CMV and suggested that low cell doses (as low as 105/kg) of polyclonal T cells can be therapeutically effective.28,30,31,32
Figure 26–1.
Scheme for adoptive T-cell therapy with antigen-specific T cells. Antigen-specific T cells can be isolated by in vitro culture, expanded in long- or short-term culture, and then transferred in large numbers to patients. Alternatively, antigen-specific T cells can be isolated using direct methods and transferred immediately to the patient at low numbers. Patients are monitored after each cell infusion for toxicity, T-cell persistence, and efficacy.
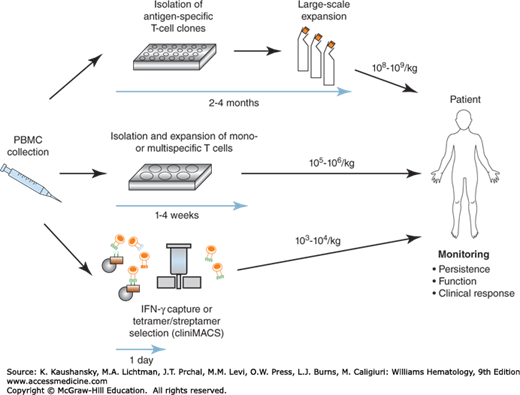
A key requirement for T-cell therapy for rapidly progressing and/or life-threatening CMV infections is to further reduce the time needed to generate the CMV-specific T cells for adoptive transfer and to improve the feasibility of this approach (see Fig. 26–1). Techniques have been developed for rapidly isolating antigen-specific T cells directly from the blood using conventional human leukocyte antigen (HLA) multimers or reversible streptamers that are comprised of soluble HLA molecules folded with the viral cognate peptide and bind T cells based on T-cell receptor (TCR) specificity.33,34,35,36 Alternatively, mAbs and immunomagnetic bead-selection strategies have been developed to capture T cells that produce interferon (IFN)-γ or have upregulated activation markers in response to antigen stimulation.37,38,39 A pilot study in 18 patients showed that CMV-specific T cells (mean cell dose 2.1 × 104/kg) isolated by IFN-γ capture techniques and transferred after brief ex vivo culture had a clinical effect.40 CMV-specific T cells were also purified from the blood of HSCT donors using HLA-peptide tetramers or reversible streptamers, transferred directly to the patients at low cell doses, and mediated antiviral activity.35,36
Transferred T cells must persist as functional memory T cells and migrate to sites of virus replication to be effective. Therefore, it was essential to verify the presence of the transferred T cells in the blood based on functional or structural properties. In the initial studies of adoptive therapy with CD8+ CMV-specific T-cell clones, assays of cytolytic activity provided an indirect semiquantitative functional measure for the presence of the transferred immune effectors.27 New approaches that use flow cytometric analysis of blood samples have been developed, such as staining with HLA multimers or intracellular staining, to detect cytokines produced after antigen stimulation. These techniques are being employed to enumerate and analyze the function of cells on a single-cell level.11,34 The unique DNA sequences of the rearranged TCR Vα or Vβ genes have also been used to evaluate survival of transferred T cells in the first trial of CMV-specific T-cell therapy.27 Quantitative real-time polymerase chain reaction with TCR-specific primers that flank the unique complementarity determining region 3 (CDR3) sequence can provide precise quantitation of transferred T cells in blood samples. Advances in high-throughput sequencing and computational analysis of the TCR sequences have now enabled much more sensitive analysis and sequencing of the CDR3 and detection of unique sequences in transferred T cells.36,41,42
The pool of memory T cells contains both CD45RO+CD62L+ central memory (TCM), and CD62L– effector memory (TEM) subsets that differ in phenotype, function, and migration.43,44,45 Studies in animal models have revealed profound differences in the ability of adoptively transferred T cells from distinct subsets to persist in vivo and revert to the memory pool. The transfer of effector cells derived from CD8+CD62L+ TCM or a rare subset of CD62L+ cells termed memory stem cells (TSCM) that share cell-surface markers of both naïve T cell (TN) and TCM cells, displayed superior survival in vivo, and/or mediated superior antitumor activity compared with more differentiated TEM.46,47,48,49 Studies in mice provide increasing evidence for a progressive differentiation model of T-cell subsets. Fate mapping studies and single-cell transfer experiments show that naïve T cells differentiate into memory and effector subsets (Fig. 26–2), and demonstrated that the stem cell–like properties of self-renewal and differentiation are present in CD62L+ TCM cells.50,51,52 It is uncertain whether the intermediate phenotype TSCM has self-renewal capability or represents a cell that is of sufficient frequency to be reliably isolated for immunotherapy. Nevertheless, these findings suggest that selecting less differentiated memory subsets as a starting population for clinical adoptive T-cell therapy can allow very low numbers of T cells to be effective, and this concept has been validated in animal models and patients with CMV infection.53 Given the considerable potential of this approach, methods are now being developed for clinically applicable serial selection strategies for human TCM subsets.54

EBV is a ubiquitous γ-herpesvirus (Chap. 82) that persists in immunocompetent hosts lifelong without causing disease. Some latently infected B cells express only the EBNA-1 protein, which has glycine-alanine repeats that inhibit its translation and processing for presentation to CD8+ T cells.55 Infected B cells may activate the latency III program of viral genes that includes EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, LMP-1, LMP-2A, and LMP-2B, and induces cell proliferation.56 In normal hosts, both the CD8+ CTL and CD4+ Th cell response to EBV infection is mainly directed against lytic viral proteins and the EBNA-3A, -3B, and -3C latency proteins.57,58 The CD4+ Th cell response may also contribute to eliminating class II MHC+ EBV-infected cells in vivo58 and EBV-specific CD8+ and CD4+ T cells cooperate to prevent the outgrowth of EBV+ B cells in immunocompetent hosts.59,60 Thus, tumors comprised of proliferating EBV+ B cells can arise in individuals with a T-cell deficiency, such as solid-organ or HSCT patients receiving intense immunosuppression, especially if T-cell depletion is used as part of the conditioning regimen.61,62 Historically, patients with EBV-induced lymphoproliferative disease (EBV-LPD) had a grave prognosis, responding poorly to antiviral drugs or chemotherapy, although early detection of EBV reactivation and treatment with mAbs specific for CD20 have improved outcomes.63 Ultimately, restoration of EBV-specific T cells is necessary for control of the virus.
The efficacy of T-cell therapy for EBV-LPD was first demonstrated in a study in which a low dose of unselected donor lymphocytes was administered to five patients with EBV-LPD after T-cell–depleted allogeneic HSCT. Complete resolution of EBV-LPD was achieved in all patients,64 but this was complicated by GVHD and two patients developed a fatal respiratory failure, demonstrating the importance of selecting EBV-specific T cells for therapy.64 The adoptive transfer of donor EBV-specific T-cell lines derived by in vitro culture with EBV-transformed lymphoblastoid cell lines (LCLs) was effective in 2 of 3 HSCT recipients with established EBV-LPD without causing GVHD.65 One patient had progressive LPD with a mutation in the EBNA-3B gene that eliminated the region encoding the epitopes targeted by the CTL line.66 To diminish the probability of escape variants, subsequent studies administered donor EBV-specific T-cell lines prophylactically to a cohort of patients at risk for EBV-LPD after T-cell–depleted allogeneic HSCT.67 No GVHD was observed, and there were no cases of LPD, although this was expected to occur in 14 percent of the patients.67 A 9-year followup report summarized the results in a total of 114 HSCT recipients, 101 of which safely received prophylactic infusions of EBV-specific CTLs.68 None of the 101 patients progressed to EBV-LPD and 11 of 13 patients with LPD achieved sustained complete remissions.68 Thus, transfer of EBV-specific T cells safely reconstituted immunity, mediated antiviral activity, and protected the majority of patients from EBV-LPD.
Severe infections with a broad array of viruses remain a serious problem for immunocompromised patients.2,3,4,5,7,69 In addition to CMV and EBV, infections with adenovirus, BK virus, human herpes virus-6 (HHV-6), herpes simplex virus (HSV), and/or varicella-zoster virus can pose a serious problem. Antiviral drugs may benefit a subset of patients, but the effects are often limited and accompanied by toxicities.70 There is inferential evidence that restoration of T-cell immunity is also critical for protection against these infections, suggesting that multivirus-specific T-cell products would have greater utility.71 EBV-LCLs that had been transfected with a recombinant adenovirus that encoded CMVpp65 were used to stimulate donor-derived PBMC and simultaneously expand T cells specific for adenovirus, CMV, and EBV.31,72,73 The infusion of such multispecific T cells into HSCT recipients augmented T-cell responses to all three viruses and promoted virus clearance. These initial reports of adoptive T-cell therapy with “broad-spectrum” T cells to treat the multiplicity of distinct viral infections that may complicate the clinical outcome of HSCT are encouraging. Studies have further extended this work and demonstrated the feasibility and clinical utility of rapidly generated T-cell lines that recognize 12 immunogenic antigens from five viruses, including CMV, EBV, adenovirus, BK virus, and HHV-6. Adoptive transfer of these T cells to 11 allogeneic transplant recipients produced a greater than 90 percent sustained virologic and clinical response.74
ADOPTIVE CELLULAR THERAPY OF MALIGNANCY
There is evidence from murine models that the host immune system has a dynamic relationship with a developing tumor and can recognize, control, and even eliminate cancer.75,76,77 Immunogenic proteins in human tumors have now been identified by screening of tumor complementary DNA (cDNA) libraries with tumor-specific T cells isolated from the blood or tumor environment,78 or by screening of patient sera for antibody responses to tumor-associated proteins.79 Distinct categories of tumor antigens have been uncovered, and several are being investigated as targets for T-cell therapy or vaccination (Table 26–1). However, the clinical translation of adoptive T-cell therapy and other immunotherapeutic modalities for human cancers has proven to be more challenging than for opportunistic viral infections. This reflects many issues, including the difficulty isolating highly avid tumor-specific T cells from cancer patients, and evasion mechanisms that tumors employ to avoid immune elimination including the local recruitment of regulatory T cells (TREG) or myeloid-derived suppressor cells, loss of antigen or HLA expression, and expression or secretion of inhibitory molecules or cytokines.80,81,82,83 Additionally, a problem distinct from the results of T-cell therapy for viruses is that transferred tumor-reactive T cells persisted only transiently in most early clinical trials, even if high-dose interleukin (IL)-2 was given to support their survival.84,85,86,87,88
A. Classes of antigens for MHC-restricted T cells
B. Classes of tumor cell surface molecules for chimeric antigen receptor-modified T cells
|
The development of immune cell therapy for malignancy has focused on melanoma because target antigens have been identified and this tumor has responded to nonspecific immune therapy with IL-2,89 and on amplifying the graft-versus-leukemia (GVL) effect after allogeneic HSCT because of the evidence that donor T cells mediate tumor eradication in this setting.90,91,92 The ability to engineer T cells to have tumor specificity by introducing TCR genes that recognize tumor-associated antigens,93 or chimeric antigen receptor (CAR) genes that encodes a single chain mAb domain linked to the CD3ζ chain of the TCR, and confers recognition of a tumor-associated, cell-surface molecule, is facilitating the broader application of T-cell therapy for both human hematologic malignancies and common epithelial cancers.94,95,96,97
Early studies demonstrated that the adoptive transfer of autologous polyclonal tumor-infiltrating lymphocytes (TILs), isolated and expanded from resected melanoma specimens, combined with the administration of high-dose IL-2 resulted in a 31 percent response rate in patients with advanced melanoma.98 Most of the responses were transient, but these results validated the potential to eradicate a human solid tumor with immunotherapy. These results also encouraged efforts to define the antigens recognized by TILs in responding patients, and to refine the approaches to augmenting T-cell responses to tumor antigens.
Melanoma has served as a model for the discovery of human tumor antigens because T cells specific for melanoma cells can often be detected in the blood or the tumor microenvironment. A landmark in cancer immunotherapy was the identification by cDNA expression cloning of MAGE-1, which is a member of the cancer-testes antigen class of tumor associated antigens.78 Several additional shared tumor/self-melanocyte differentiation antigens recognized by CD8+ and/or CD4+ T cells have been discovered, including differentiation proteins that function in normal melanocyte physiology such as tyrosinase, gp100, and MART-1; and other cancer testis antigens such as NY-ESO-1.78,99,100 Melanosome antigens are also expressed in normal tissues (skin, retina), and toxicity because of autoimmunity is a concern. Mutated proteins that arise as a consequence of the genetic instability of tumors are being identified as critical targets of immune recognition, and these offer the greatest promise for selectively targeting tumor cells without recognition of normal cells.101
The adoptive transfer of tumor-specific T-cell clones or oligoclonal populations of T cells expanded ex vivo can, in principle, allow control over the magnitude and function of the tumor-reactive T-cell response in the patient. If the tumor is easily accessible, tumor-reactive T cells can sometimes be isolated directly from the tumor biopsies by culture in high-dose IL-2.102 Melanoma-reactive T cells can also be isolated using autologous dendritic cells pulsed with synthetic peptide antigen, but this approach can enrich low-avidity T cells that have a limited capacity to persist and function in vivo.103
Initial clinical trials of T-cell therapy for melanoma employed CD8+ T-cell clones specific for MART-1 or gp100; or clonal or polyclonal melanoma-reactive T cells derived and expanded from TILs.84,104,105 The transferred T-cell clones given with low-dose IL-2 mediated transient antitumor activity in some patients with advanced disease, but did not persist for long term.104 The response rate was higher in patients treated with polyclonal TILs and high-dose IL-2.105 T-cell persistence was highly variable and only rarely sustained despite the infusion of large T-cell numbers (up to 1011).84,104 The inability of T cells to persist in vivo could reflect an inadequate antigen-specific CD4+ Th response, terminal differentiation of T cells during expansion, activation-induced T-cell death at the tumor site, or cell death as a consequence of IL-2 withdrawal.84,106
A major advance in the field was the demonstration that the transferred human T-cell persistence and therapeutic efficacy could be improved by pretreatment of the patient with a lymphodepleting regimen containing cyclophosphamide and fludarabine.107,108 In these studies, high-dose IL-2 was administered daily after TIL transfer until toxicity required it be discontinued. A subset of the patients achieved prolonged high-level engraftment of one or a few tumor-reactive CD8+ T-cell clonotypes present in the infused polyclonal T-cell product.107,108,109,110 In a followup analysis of a large cohort of patients, the overall and complete response rates were approximately 50 percent and 20 percent, respectively.108,111 The antitumor activity correlated with the persistence of high levels of transferred tumor-reactive CD8+ T cells. Several mechanisms make the lymphopenic environment favorable for T-cell transfer, including less competition for homeostatic cytokines such as IL-15 and IL-7 that promote lymphocyte survival,112,113 and the elimination of CD4+CD25+ TREG cells.114 Studies in murine models have confirmed that severe lymphodepletion can be exploited to improve the antitumor efficacy of the transferred T cells.115 The addition of 2 Gy or 12 Gy of total-body irradiation to the lymphodepleting treatment with cyclophosphamide and fludarabine before TIL transfer increased the response rate to 52 percent and 72 percent, respectively.116 The results of TIL therapy in a metastatic tumor that is unresponsive to conventional therapy were achieved with moderate toxicity, demonstrating the encouraging potential of this therapy.
Studies of the mechanisms of tumor eradication in melanoma are providing insights for treatment of other cancers with T-cell therapy. Exome sequencing of melanoma has shown that the frequency of mutational events is high,101,117 and detailed analysis of the specificity of TILs showed that in addition to T cells specific for shared tumor/self-melanocyte differentiation antigens,118 such mutated gene products encoded neoepitopes that were often targets of immune recognition.101,108,119 The identification of neoepitope T cells in nonmelanoma cancers is of considerable interest because mutations in other tumors may be similarly targeted. In one example, TIL therapy was used to successfully treat a patient with metastatic cholangiocarcinoma.120 Whole-exome sequencing of the tumor identified a mutation within the erbb2 interacting protein (ERBB2IP) that was recognized by a subset of CD4+ T cells in the TIL product. Treatment with a greater than 95 percent pure population of mutation-reactive T cells resulted in dramatic and durable tumor regression in this patient.120 These results illustrate the potential of patient specific T-cell therapy targeting immunogenic mutations present in their tumor and highlight the need to apply advanced genomic technologies to the discovery of targets for immune therapy in cancer.108,120
Collectively, significant progress has been made in cellular therapy for melanoma, but additional studies are necessary to define the optimal and safest regimens for adoptive therapy with tumor-reactive T cells. Advances in our understanding of the role of individual cytokines in T-cell survival in vitro and in vivo, or the regulation of T-cell activation and homeostasis will provide new opportunities for improving the persistence of in vitro expanded T cells after transfer, perhaps obviating use of toxic chemoradiotherapy to deplete lymphocytes before T-cell infusions. Most of the initial efforts have focused on the CD8+ T-cell response to tumor antigens, but newer data highlights the potential of tumor-specific CD4+ T cells. Combining T-cell therapy with targeted depletion of TREG, checkpoint inhibitors, and vaccines is also under investigation, and may help overcome mechanisms by which tumors evade elimination by limiting the quantity and quality of the host response.
Allogeneic donor T cells contained in or derived from the stem cell graft can mount a GVL effect that can contribute to the eradication of hematologic cancers, including leukemia.90
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


