INTRODUCTION
SUMMARY
Apoptosis was originally coined to describe the morphologic features of a form of cell death characterized by cell shrinkage, membrane blebbing, and nuclear condensation. This type of cell death occurs in a wide variety of physiologic contexts, and thus is sometimes referred to as programmed cell death. Apoptosis occurs in all animal species as a means to balance cell proliferation with cell loss. The physiologic benefits of apoptosis include eliminating cells that are unneeded, defective, or infected, and maintenance of tissue homeostasis by continuously renewing adult tissues so as to maintain appropriate organ mass. In the hematopoietic system, production of leukocytes is delicately balanced against cell death, until a need arises for rapidly generating immune and inflammatory cells for combating pathogens. The life span of hematopoietic cells is regulated by numerous cytokines and lymphokines, as well as by signals derived from microanatomical niches through cell adhesion molecules and other regulators. Defects in the regulation of hematopoietic cell life span contribute to myriad diseases, including disorders characterized by inappropriate cell accumulation, such as leukemia, lymphoma, and autoimmunity, and diseases where pathologic loss of cells occurs, such as immunodeficiency and various blood dyscrasias.
Acronyms and Abbreviations
ALL, acute lymphocytic leukemia; ALPS, autoimmune lymphoproliferative syndrome; Asp, aspartic acid; B-CLL, B-cell chronic lymphocytic leukemia; BH, Bcl-2 homology domain; CARDs, caspase recruitment domains; caspases, cysteine aspartyl proteases; CLLs, chronic lymphocytic leukemias; CHOP, C/EBP homologous protein; CML, chronic myelogenous leukemias; CTL, cytolytic T lymphocyte; Cyt-c, cytochrome c; DD, death domain; DEDs, death effector domains; DISC, death-inducing signaling complex; DLBCL, diffuse large B-cell lymphoma; DR, death receptor; EBV, Epstein-Barr virus; ER, endoplasmic reticulum; FasL, Fas ligand; FKHD, forkhead transcription factors; IAP, inhibitor of apoptosis; IBD, inflammatory bowel disease; IgH, immunoglobulin heavy chain; IKKs, I-κB kinases; IL, interleukin; KSV, Kaposi sarcoma virus; MALT, mucosa-associated lymphoid tissue; miRNAs, microRNAs; MLKL, mixed-lineage kinase domain-like; MMs, multiple myelomas; MOMP, mitochondrial outer membrane permeabilization; MPT, mitochondrial permeability transition; NHLs, non-Hodgkin lymphomas; NK, natural killer; PARP, poly-ADP ribosyl polymerase; PCD, programmed cell death; PI3K, phosphatidylinositol 3’-kinase; pro/pre–B-cells, B-lymphocyte progenitors; ROS, reactive oxygen species; TNF, tumor necrosis factor; TNFR1, TNF receptor-1; UBCs, ubiquitin conjugating enzymes.
It is now well established that defects in the normal mechanisms that control programmed cell death (PCD) occur commonly in human diseases. Cell numbers in the body are governed not only by cell division, which determines the rate of cell production, but also by cell death, which dictates the rate of cell loss. In the course of a typical day, an average adult human produces, and in parallel eradicates, approximately 50 to 70 billion cells, representing approximately 1 million cells per second. Normally, these two processes of cell division and cell death are tightly coupled so that no net increase in cell numbers occurs, or so thatsuch increases represent only temporary responses to environmental stimuli. However, alternations in the expression or function of the genes that control PCD can upset this delicate balance, contributing to or causing disease.
In most cases, PCD occurs by apoptosis. Apoptosis is defined by its morphologic features. As viewed with the assistance of the light- (or, preferably, electron-) microscope, the characteristics of the apoptotic cell include chromatin condensation and nuclear fragmentation (pyknosis), plasma membrane blebbing, and cell shrinkage. Eventually, the cell breaks into small membrane-surrounded fragments (apoptotic bodies), which are cleared by phagocytosis, without inciting an inflammatory response. The release of apoptotic bodies is what inspired the term “apoptosis” from the Greek, meaning “to fall away from” and conjuring notions of the falling of leaves in the autumn from deciduous trees.1
In recent years, the molecular machinery responsible for apoptosis has been elucidated, revealing a family of intracellular proteases, caspases (cysteine aspartyl proteases), which are responsible directly or indirectly for most of the morphologic and biochemical changes that characterize the phenomenon of apoptosis.2,3 Diverse regulators of the caspases have also been discovered, including activators and inhibitors of these cell death proteases. Inputs from signal transduction pathways into the core of the cell death machinery have also been identified, demonstrating ways of linking environmental stimuli to cell death responses or cell survival maintenance. Knowledge of the molecular mechanisms of apoptosis is providing insights into the pathogenesis of many diseases, revealing strategies for possible novel treatments.
CASPASES—PROTEASES THAT CAUSE APOPTOSIS
Intracellular proteases called caspases are responsible for most of the morphologic changes that we recognize as “apoptosis,” as well as many of the biochemical changes often associated with this route of cell demise. Specifically, activation of a family of intracellular cysteine proteases that cleave their substrates at aspartic acid residues, known as “caspases” for cysteine aspartyl-specific proteases.4 These proteases are present as inactive zymogens in essentially all animal cells, but can be triggered to assume active states, generally involving their proteolytic processing at conserved aspartic acid (Asp) residues. During activation, the zymogen proproteins are cleaved to generate the large (~20 kDa) and small (~10 kDa) subunits of the active enzymes, typically liberating an N-terminal prodomain from the processed polypeptide chain. The active enzymes consist of heterotetramers composed of two large and two small subunits, generally with two active sites per molecule.2,3
The observation that caspases cleave their substrates at Asp residues and are also activated by proteolytic processing at Asp residues makes evident that these proteases collaborate in proteolytic cascades, where caspases activate themselves and each other. Humans contain 11 caspases. They can be subgrouped according to either their amino-acid sequence similarities or their protease specificities.
From a functional perceptive, it is useful to view the caspases as either upstream “initiator” caspases or downstream “effector” caspases.5 The zymogen forms of upstream initiator caspases possess large N-terminal prodomains, which function as protein interaction modules, allowing them to interact with various proteins that trigger caspase activation. In contrast, the proforms of downstream effector caspases contain only short N-terminal prodomains, serving no apparent function. Downstream caspases are largely dependent on upstream caspases for their proteolytic processing and activation. The substrates of effector caspases are myriad, as revealed in recent years by unbiased proteomics approaches. Substrates include cytoskeletal and nuclear matrix proteins, chromatin-modifying (e.g., poly-ADP ribosyl polymerase [PARP]) and DNA repair proteins, inhibitory subunits of endonucleases (CIDE-family proteins), protein kinases (often separating the autorepressing regulatory domains from catalytic domains) and other signal transduction proteins.
CASPASE ACTIVATION PATHWAYS
Several pathways for activating caspases have been delineated (Fig. 15–1). The simplest is exploited by cytolytic T lymphocytes (CTLs) and natural killer (NK) cells, which introduce apoptosis-inducing proteases, particularly granzyme B (a serine protease), into effective intracellular compartments of target cells via perforin-dependent mechanisms.6 Unlike the caspases, granzyme B is a serine protease. However, similar to the caspases, granzyme B specifically cleaves its substrates at Asp residues. Granzyme B is capable of cleaving and activating multiple caspases and some caspase substrates.7 Endogenous and viral inhibitors of granzyme B have been identified, accounting for resistance to this apoptotic inducer.8,9,10
Figure 15–1.
Pathways for caspase activation. The major pathways for caspase activation in mammalian cells are presented. The extrinsic (left, upper) is induced by members of the tumor necrosis factor (TNF) family of cytokine receptors such as TNF receptor-1 (TNFR1), Fas, and the tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) receptors. These proteins recruit adapter proteins to their cytosolic death domains (DDs), including the Fas-associated death domain (FADD), which then bind the death effector domain (DED)–containing procaspases, particularly procaspase-8, inducing their activation. Cytolytic T lymphocytes (CTLs) and natural killer (NK) cells introduce the protease granzyme B into target cells (right, upper). This protease cleaves and activates multiple members of the caspase family. The intrinsic pathway (left, lower) is initiated by release of cytochrome c from mitochondria, induced by various stimuli, including elevations in the levels of pore-forming proapoptotic Bcl-2 family proteins, such as Bax and Bak. In the cytosol, cytochrome c binds and activates Apaf-1, allowing it to associate with and activate procaspase-9. Active caspase-9 (intrinsic) and caspase-8 (extrinsic) have been shown to directly cleave and activate the effector protease, caspase-3. Because other caspases also become involved in these pathways (not shown), the schematic represents an oversimplification of the events that occur in vivo. Additionally, disturbances in the function of the endoplasmic reticulum (ER) are also linked to the intrinsic and extrinsic apoptosis pathways, through both Ca2+ transfer to mitochondria and via transcriptional mechanisms that include induction of CHOP expression, which, in turn, stimulates expression of death receptor 5 (DR5) and Bim (proapoptotic Bcl-2 family member) (right, lower). Rectangles indicate proapoptotic proteins whereas ellipses indicate antiapoptotic proteins.
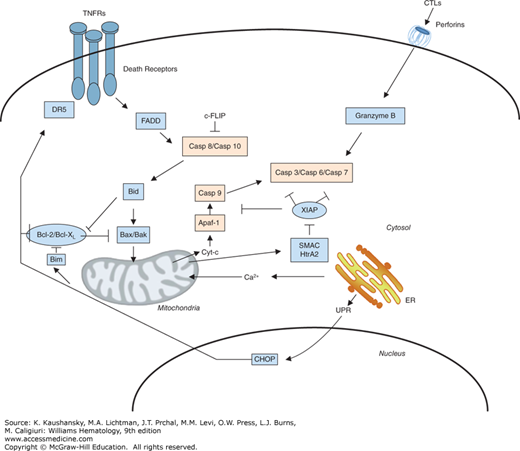
Another caspase-activation pathway is represented by tumor necrosis factor (TNF) family receptors. Eight of the approximately 30 known members of the TNF family in humans contain a so-called death domain (DD) in their cytosolic tails.11 Several of these DD-containing TNF family receptors use caspase activation as a signaling mechanism, including TNF receptor-1 (TNFR1)/CD120a; Fas/APO1/CD95; death receptor (DR)-3 (DR3)/Apo2/Weasle; DR4/ tumor necrosis factor–related apoptosis-inducing ligand receptor 1 (TRAILR1); DR5/TRAILR2; and DR6. Ligation of these receptors at the cell surface results in receptor clustering and recruitment of several intracellular proteins, including certain procaspases, to the cytosolic domains of these receptors, forming a “death-inducing signaling complex” that triggers caspase activation and leads to apoptosis.12,13
The specific caspases summoned to the DISC are caspase-8 and, in some cases, caspase-10. These caspases contain so-called death effector domains (DEDs) in their N-terminal prodomains that bind to a corresponding DED in the Fas-associated death domain (FADD), a bipartite adapter protein containing a DD and a DED. FADD functions as a molecular bridge between the DD and DED domain families, and is, in fact, the only protein in the human genome with this dual domain structure. Consequently, cells from mice in which the fadd gene has been knocked out are resistant to apoptosis induction by TNF family cytokines and their receptors. Cells derived from caspase-8 knockout mice also fail to undergo apoptosis in response to ligands or antibodies that activate TNF family DRs, demonstrating an essential role for this caspase in this pathway.14 However, mice lack the highly homologous protease, caspase-10, which is found in humans, having arisen from an apparent gene duplication on chromosome 2.15 Thus, caspases 8 and 10 may play redundant roles in human cells.
Mitochondria also play important roles in apoptosis, releasing cytochrome c (Cyt-c) into the cytosol, which then causes assembly of a multiprotein caspase-activating complex, referred to as the “apoptosome.”16,17 The central component of the apoptosome is Apaf1, a caspase-activating protein that oligomerizes upon binding Cyt-c and which specifically binds procaspase-9. Apaf1 and procaspase-9 interact with each other via their caspase recruitment domains (CARDs). Such CARD–CARD interactions play important roles in many steps in apoptosis pathways. In addition to Cyt-c, mitochondria also release several other proteins of relevance to apoptosis, including endonuclease G, AIF (an activator of nuclear endonucleases), and SMAC (Diablo) and Omi (HtrA), antagonists of a family of caspase-inhibitory proteins known as the IAPs (inhibitors of apoptosis) (see section “Inhibitors of Apoptosis” below).
The central importance of the Cyt-c–dependent pathway for apoptosis is underscored by the observation that cells derived from mice in which either the apaf1 or procaspase-9 genes have been ablated are incapable of undergoing apoptosis in response to agents that trigger Cyt-c release from mitochondria.18 Nevertheless, such cells can die by nonapoptotic routes,19 demonstrating that mitochondria control both caspase-dependent and caspase-independent cell death pathways. Moreover, distinguishing mitochondria-driven apoptotic from nonapoptotic cell death can be challenging in many contexts because of the similar morphologic features caused probably by some of the proteins released from these organelle such as endonuclease G and AIF, which promote chromatin condensation and DNA fragmentation. The mitochondrial mechanisms for apoptotic and nonapoptotic cell death are activated by myriad stimuli, including growth factor deprivation, oxidants, Ca2+ overload, DNA-damaging agents, microtubule-modifying drugs, and much more.17,20 In this sense, mitochondria are sometimes viewed as central integrators of cell stress signals that dictate ultimately cell life and death decisions.
Mitochondria can also participate in cell death pathways induced via TNF family DRs, through crosstalk mechanisms involving proteins such as Bid, BAR, and Bap31.21,22,23,24 However, mitochondrial (“intrinsic”) and DR (“extrinsic”) pathways for caspase activation are fully capable of independent operation in most types of cells.25
Cell death mechanisms are also linked to endoplasmic reticulum (ER). In most cases, however, these ER-initiated signals ultimately seem to impinge on mitochondria as the downstream effectors of the cell death pathway. In this regard, the ER is a central regulator of intracellular Ca2+, and ER membranes form close contacts with mitochondria to create structures where Ca2+ effluxes from ER into mitochondria, thereby impacting mitochondrial function in profound ways that can either promote cell life or cause death. Too much Ca2+ entry into mitochondria, for instance, triggers a phenomenon called mitochondrial permeability transition (MPT) in which the organelles swell and eventually rupture.
However, in addition to the role of ER Ca2+ and mitochondria-driven cell death, another pathway for apoptosis has been linked to accumulation of unfolded proteins in the ER. Specifically, ER stress induces expression of the proapoptotic transcription factor CHOP, which, in turn, stimulates expression of DR5 (TRAILR2), causing caspase-8-dependent apoptosis.26 Additionally, CHOP has been reported to directly stimulate transcription of the gene encoding Bim, a proapoptotic member of the Bcl-2 family (see section “Suppressors of Apoptosis” below) that stimulates Cyt-c release from mitochondria. Thus, ER stress has multiple potential routes of stimulating cell death pathways, with the predominant pathway probably varying among cell types and pathophysiologic contexts.
Although diverse mechanisms exist for activating initiator Caspases, as outlined above, in most instances, the biochemical mechanisms appear to be remarkably similar. Much of caspase activation and can be explained by the “induced proximity model,”27 in which forcing dimerization of procaspases results in conformational states that promote protease activation, typically resulting in cleavage events that lock the proteases into their fully active state. This mechanism is clearly operative in the caspase-activation pathways induced by TNF family receptors (extrinsic pathway) and Cyt-c/mitochondria (“intrinsic pathway”).
SUPPRESSORS OF APOPTOSIS
Given the critical importance of making the correct choices about cell life–death decisions in complex multicellular organisms, it is not surprising that the pathways governing caspase activation are under exquisite control by networks of proteins that directly or indirectly communicate with these proteases. A delicate balance between proapoptotic and antiapoptotic regulators of apoptosis pathways is at play on a continual basis, ensuring the survival of long-lived cells and the proper turnover of short-lived cells in a variety of tissues, including the marrow, thymus, and peripheral lymphoid tissues. The antiapoptotic proteins responsible for creating roadblocks to cell death have been mapped to specific caspase-activation pathways.
The Bcl-2 family represents a large group of proteins (number >26 in humans) that control mitochondria-dependent steps in cell-death pathways, including dictating whether Cyt-c is or is not released from these organelles (Fig. 15–2). Both proapoptotic and antiapoptotic Bcl-2 family proteins have been delineated.28 These proteins are best known for their roles in controlling the intrinsic (mitochondrial) cell-death pathway,20 although effects on the ER-pathway for cell death have also been documented.29 Even though the human genome encodes at least 26 Bcl-2 family proteins, only six of these are antiapoptotic (in humans, Bcl-2, Bcl-XL [BCL2L1], Bcl-W [BCL2L2], Mcl-1 [BCL2L3], Bfl-1 [BCL2L5], and Bcl-B [BCL2L10]). Several types of animal viruses also harbor Bcl-2 family genes within their genomes, including herpes viruses implicated in cancer such as the Epstein-Barr virus (EBV) and Kaposi sarcoma virus (KSV). The relative ratios of anti- and proapoptotic Bcl-2 family proteins dictate the ultimate sensitivity or resistance of cells to various apoptotic stimuli, including growth factor deprivation, hypoxia, radiation, anticancer drugs, oxidants, and Ca2+ overload.
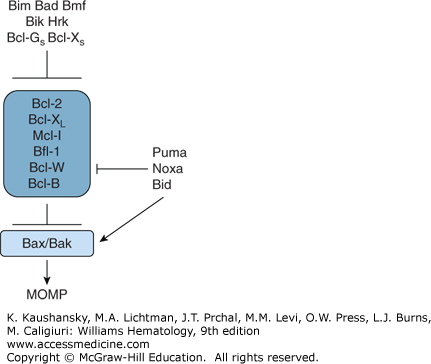
Various Bcl-2 family members play important roles in controlling the life spans of hematopoietic cells, as evidenced by phenotypes generated in genetically engineered mice (gene knockouts and transgenics) and also (in some cases) by human clinical experiences with experimental therapeutics targeting some of these proteins. For example, antiapoptotic protein Bcl-2 is required for survival of mature T cells and B cells, with deficiency of Bcl-2 causing lymphopenia. Conversely, the proapoptotic protein Bim is necessary for limiting expansion of T and B lymphocytes, with deficiency of Bim causing lymphocytosis. Bim also plays important role in eradicating autoreactive T cells in the thymus (“negative selection”), having important implications for mechanisms of autoimmune diseases.30 Bcl-XL is required for platelet homeostasis, such that either genetic or pharmacologically induced Bcl-XL deficiency causes thrombocytopenia. Antiapoptotic protein Mcl-1 is particularly important for survival of the myeloid lineage in mice, as well as contributing to lymphocyte survival. Conversely, antiapoptotic protein Bcl-W is not required for hematopoiesis in mice, despite being widely expressed in myeloid lineage cells. It should be noted that direct comparisons of gene manipulations in mice with the human circumstance are not always possible because of genomic differences in the Bcl-2 family genes of mice versus humans (e.g., human Bfl-1 versus murine A1; human Bcl-B versus murine Boo/Diva).
Many members of the Bcl-2 family have a hydrophobic stretch of amino acids near their carboxyl-terminus that anchors them in the outer mitochondrial membrane.17 In contrast, other Bcl-2 family members such as Bid, Bim, and Bad, lack these membrane-anchoring domains, but dynamically target mitochondria in response to specific stimuli. Still others have the membrane-anchoring domain but keep it latched against the body of the protein until stimulated to expose it (e.g., Bax).31
Based on their predicted (or experimentally determined) three-dimensional structures, Bcl-2 family proteins can be broadly divided into two groups. One subset of these proteins is probably similar in structure to the pore-forming domains of bacterial toxins, such as the colicins and diphtheria toxin.32,33,34,35 These α-helical pore-like proteins include both antiapoptotic proteins (Bcl-2, Bcl-XL, Mcl-1, Bfl-1, Bcl-W, Bcl-B) and proapoptotic proteins (Bax, Bak, Bok, and Bid). Most of the proteins in this subcategory can be recognized by conserved stretches of amino acid sequence homology, including the presence of Bcl-2 homology (BH) domains, BH1, BH2, BH3, and sometimes BH4. However, this is not uniformly the case, as the Bid protein contains only a BH3 domain but has been determined to share the same overall protein-fold with Bcl-XL, Bcl-2, and Bax.33,34 Where tested to date, these proteins have all been shown to form ion-conducting channels in synthetic membranes in vitro, including Bcl-2, Bcl-XL, Bax, and Bid,36,37,38,39,40 but the significance of this pore activity remains unclear.
The other subset of Bcl-2 family proteins appears to have in common only the presence of the BH3 domain, including Bad, Bik, Bim, Hrk, Bcl-GS, p193, APR (Noxa), and PUMA. These “BH3-only” proteins are uniformly proapoptotic. Their cell-death–inducing activity depends, in most cases, on their ability to dimerize with antiapoptotic Bcl-2 family members, functioning as trans-dominant inhibitors of proteins such as Bcl-2 and Bcl-XL.41,42 However, some of these proteins (e.g., Bid, PUMA, Bim) can also interact with proapoptotic proteins (e.g., Bax, Bak), functioning as agonists of the killers, in addition to dimerizing with antiapoptotics (e.g., Bcl-2; Bcl-XL) to function as antagonists of these cell-survival proteins (see Fig. 15–2).28,43 Binding of Bid to Bax or Bak promotes insertion of these proteins into membranes where they oligomerize, apparently forming large pores through which molecules such as Cyt-c, SMAC, and Omi can escape from mitochondria or causing an increase in the permeability of the outer membrane of mitochondria through more complex mechanisms.44,45
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


