“There are about 25 trillion red blood cells packed inside a single human body—line them side by side and they will circle the earth 15 times. This astoundingly large surface area was created to deliver what each cell in our body needs: oxygen.”
7.1 Anemia
Anemia is the state of reduced hemoglobin in blood.
Clinical pathophysiology: Presentation can vary with the degree of anemia—from exercise intolerance and easy fatigability to shortness of breath at rest. Also, with decrease in oxygen delivery to tissues, the body signals the heart to pump more blood, so there is increase in heart rate (palpitations) and increase in stroke volume (physical exam may reveal high flow midsystolic murmur in aortic area, known as a functional murmur). Additional exam findings includes scleral pallor.
Cardiac ischemia can develop due to increased work load (tachycardia) and increased demand for O2 while O2 content in blood is low.
In long-standing severe anemia, high output heart failure can ensue.
7.1.1 Microcytic Anemias (MCV < 80)
Low mean cell volume (MCV) means there is a problem with production of hemoglobin component of red blood cell (RBC).

Iron Deficiency Anemia
What happens to serum iron studies in iron deficiency state?
Ferritin is the storage form of iron. When the body iron stores are low, serum ferritin is low.
Low iron stores result in compensatory increase in synthesis of transferrin. (Transferrin is a serum protein that binds and transports iron to various tissues.)
Increase in transferrin means increase in serum iron binding capacity.
As serum iron is low, the iron saturation is low. Iron saturation refers to the number of iron molecules that are bound to transferrin molecule.
Serum iron studies cofirms iron defeciency state. NSIM is to treat the iron deficiency and find the cause.
Rx: Iron replacement therapy (IV or oral) and supportive transfusion as needed. Follow-up response with reticulocyte count and iron studies.
3 If there is no response to iron supplementation, 1st SIM is to check for compliance and ask for side effects. Main reason patients do not take iron pills is because of GI upset.
Etiology
Less than 50 years: likely etiology in this age group is menorrhagia in females, and peptic ulcer disease in males.
More than 50 years: consider colon cancer. Check fecal occult blood test and do colonoscopy.
Prematurity: most of the iron stores in the newborns is acquired from mother during third trimester of pregnancy.
In children, there is increased demand for iron, thus decreased intake of iron can easily result in iron deficiency.
In children, if there is no response to iron supplementation, gastrointestinal bleeding due to Meckel’s diverticulum is the potential cause.
Chronic mechanical hemolysis (e.g., in mechanical heart valve).
Increased demand after erythropoietin treatment in end-stage renal disease (ESRD) patients.
Anemia of Chronic Disease
Etiology: Any cause of chronic inflammation such as collagen vascular diseases, active malignancy, chronic infection (e.g., chronic osteomyelitis), etc.
Pathphysiology: Increase in inflammatory mediators signal liver to produce hepcidin that blocks the release of iron from its storage form. Increased iron stores mean high serum ferritin
5 Ferritin is also a marker of inflammation. It can be high in patients with ongoing infection and/or inflammation.
Management: Treat underlying cause.
Supportive Rx: In patients with Hb < 10 g/dL, iron replacement therapy is indicated if ferritin < 100 ng/mL OR iron (transferrin) saturation < 20%. Erythropoietin-stimulating agents are given (e.g., darbepoetin) after ensuring adequate iron stores.
Sideroblastic Anemia
Pathophysiology: Inadequate or abnormal synthesis of protoporphyrin to make heme. This will result in idle iron not being used for hemoglobin synthesis (so serum ferritin, serum iron, and iron saturation is high).
Vit B6 (pyridoxine) deficiency: it can be hereditary or acquired (e.g., due to isoniazid use)
Clonal sideroblastosis, likely due to underlying myelodysplastic syndrome
Diagnostic evaluation: usually history and iron study point toward the diagnosis.
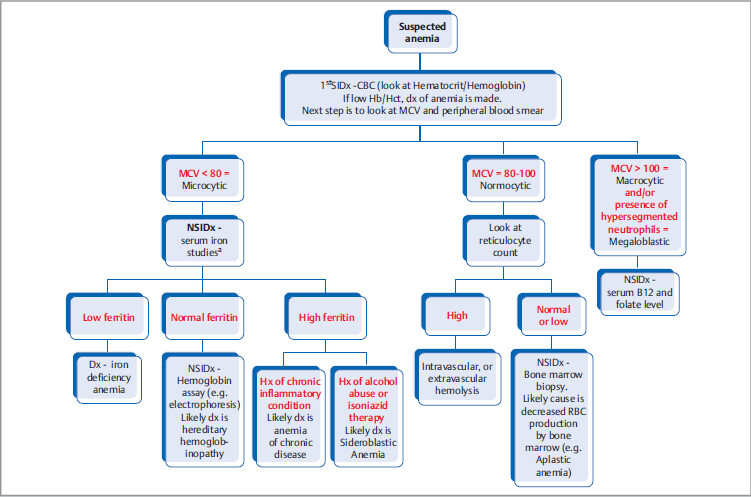
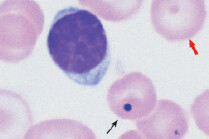
The most specific test is bone marrow biopsy with iron staining (Prussian blue stain) of specimen. In absence of protoporphyrin to complete the hemoglobin synthesis, iron dedicated for heme synthesis becomes trapped in mitochondria of erythrocyte precursors (mitochondria is where heme biosynthesis begins). This shows up like an apparent ring around the nucleus, hence called ringed sideroblasts (white arrows). Sometimes you can see stippled RBCs (basophilic stippling) on peripheral blood smear.
Some types of refractory sideroblastic anemia are associated with myelodysplastic syndrome and development of leukemia. Presence of sideroblasts along with high MCV points toward this development.
Thalassemia
Definition: Abnormal production of globin protein (of hemoglobin) due to hereditary genetic defect.
β thalassemia is genetic defect in production of β-globin protein | |
Four genes are responsible for production of α-globin, so there are four levels of severity: | There are two genes responsible for production of β-globin:
|
a Fetal hemoglobin does not have β-globin protein, so fetuses and newborns are not affected. After 6 months, adult hemoglobin replaces fetal hemoglobin. | |
Diagnostic evaluation:
When to suspect: Microcytic anemia with normal iron panel, and normal red cell distribution width (RDW). Peripheral blood smear shows homogenous hypochromic cells and target,
or tear drop cells(see ‘1’ on the right image).
6 When “target cells” (2) are mentioned in a test question, think of the following conditions: Hemoglobinopathies (hemoglobin C, sickle cell disease, thalassemias, etc.) Liver disease Splenectomy
7 FYI, Hb assaying methods include high-performance liquid chromatography, capillary electrophoresis, and isoelectric fusing. These newer methods are preferred over traditional gel electrophoresis.
Confirmatory testing includes DNA-based methods (e.g., PCR).
Hemoglobin Assay
To undestand hemoglobin assays, we need to know about different types of hemoglobin.

In alpha thalassemias, there is reduced synthesis of all forms of hemoglobin. Four gamma (γ) chains can fuse to form Bart’s hemoblogin, and percentage of Bart’s hemoblogin corresponds with severity of alpha thalassemia. In 3 or 4 genes-deleted severe alpha thalassemias, excess of β chains can fuse to form β-globin tetramer (HbH).
In beta thalassemia, there can be compensatory increase in Hb A2 or HbF.
Management
Severe anemia is treated with regular blood transfusion.
8 This can lead to secondary hemochromatosis (iron overload) later in life. Now serum iron panel will show elevated ferritin and increased iron saturation.
Splenectomy can decrease the need for transfusion. Spleen is where donor erythrocytes are naturally destroyed.
Prenatal Screening for Thalassemias
Indication: pregnant patients with abnormal screening complete blood count (CBC) (low MCV with normal RDW), or in couples with positive family hx.
Test of choice is hemoglobin assay of both parents.
If hemoglobin assay is negative in at least one parent, the risk of clinically important disease is low. Reassure the parents-to-be. If both are carriers, only then there is a risk of clinically important disease in the baby. Offer counseling and further prenatal diagnostic options.
3. Patient has hx of multiple RBC transfusions in the past. Hemoglobin assay shows increased HbA2 and HbF. What is the likely dx?
4. Patient with history of hypertension, dyslipidemia, diabetes, and rheumatoid arthritis is found to have anemia. TIBC and iron saturation is low and serum ferritin is high. What is the likely cause of anemia?
7.1.2 Macrocytic (MCV > 100) or Megaloblastic Anemias
Background: Bone marrow stem cells require vitamin B12 (cobalamin) and folic acid for DNA synthesis. Deficiency of either can result in abnormal DNA synthesis and formation of large RBCs (macroytic anemia). Antimetabolite medications, such as methotrexate, phenytoin, trimethoprim, 6-mercaptopurine, can also impair DNA synthesis resulting in macrocytosis.
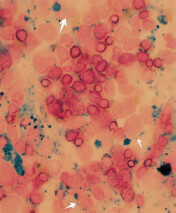
Work-up: If macrocytosis is seen, NSIDx is peripheral blood smear and serum folate and vitamin B12 level. Presence of hypersegmented neutrophils (≥ 5 lobes in nucleus) is pathognomonic of megaloblastic anemia.
Vitamin B12 Deficiency
Mechanism of vitamin B12 absorption: Vitamin B12 is released from food in presence of gastric acid. It then binds with intrinsic factor (secreted from gastric parietal cells) in the duodenum, which requires presence of pancreatic enzymes. This vitamin B12–intrinsic factor complex is absorbed by ileum into the circulation.
Etiology of vitamin B12 deficiency:
Additonal manifestation (neurological symptoms): Vitamin B12 deficiency can result in excessive production of methylmalonic acid, which is toxic to myelin sheath leading to demyelination. Patients may develop decreased position and vibratory sense. Glove stocking peripheral neuropathy is common, but note that any nervous system can be involved (autonomic neuropathy, motor paralysis, cranial nerves involvement, dementia, etc.)
Diagnostic Evaluation
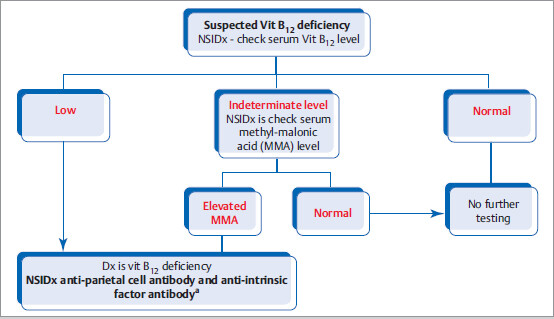
Treatment: Oral or intramuscular vitamin B12 supplementation. In patients with significant anemia, neurological symptoms, or pernicious anemia, start intramuscular vitamin B12 injections.
10 Parenteral vitamin B12 treatment can result in hypokalemia due to rapid uptake of potassium by newly forming hematopoietic cells.
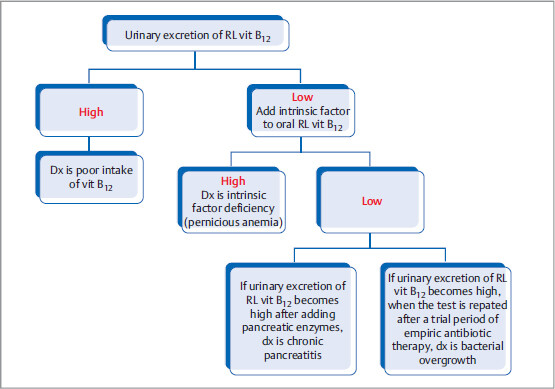
Schilling Test
Even though it is not used for diagnosis nowadays, it is asked on exam, as it requires knowledge of mechanism of vitamin B12 absorption.
How it is performed: Intramuscular vitamin B12 is first given to saturate all the serum vitamin B12 receptors. Next step is oral administration of radiolabeled (RL) vitamin B12. If absorption is intact, the absorbed RL vitamin B12 will be directly excreted with urine (as there are no seats left to bind in receptors).
If urinary excretion of RL (radio-labeled) vitamin B12 is high, then the deficiency is related to poor intake.
If urinary excretion of RL vitamin B12 is low, then NSIDx is to add intrinsic factor to oral RL vitamin B12.
If urinary excretion is high, dx of intrinsic factor deficiency is made.
If urinary excretion is still low, then other dx that can be considered are pancreatitis (test normalizes after pancreatic enzyme supplementation), bacterial overgrowth (normalizes when the test is repated after a trial period of empiric antibiotic therapy), etc.
Folate Deficiency
Treatment: Replace folate (almost always orally, because the MCC of folate deficiency is due to decreased intake or absorption).
Vitamin B12 and Folate Deficiency
Homocysteine can be elevated in both.
Both can cause mild hemolysis (elevated lactate dehydrogenase [LDH] and elevated bilirubin) and pancytopenia, if severe. (The abnormally large blood cells get destroyed).
Only vitamin B12 deficiency can have elevated methymalonic acid level and neurological manifestation.
7.1.3 Normocytic Anemia (MCV 80–100)
Normal MCV means that there is no problem in hemoglobin or DNA synthesis. This anemia can be due to the following:
7.1.4 Hemolytic Anemia
Diagnostic work-up: To find out whether RBCs are being destroyed in the blood vessels (intravascular hemolysis) or the spleen (extravascular hemolysis), do the following tests: peripheral blood smear, Coomb’s test, serum LDH, haptoglobin, and bilirubin levels.
Lowa | ||
Can be presenta | ||
Indirect bilirubinb | ||
aWhen RBCs are destroyed intravascularly, hemoglobin is released directly into blood stream. There is a protein named haptoglobin that binds to free hemoglobin in blood and transports it to spleen. Low haptoglobin levels are indicative of intravascular hemolysis. In severe acute hemolysis, there is not enough haptoglobin to sequester excessive hemoglogbin, which ends up being filtered by kidneys (hemoglobinuria). This can be toxic to renal tubular cells, causing acute tubular necrosis and renal failure. Moreover, some of this hemoglobin gets oxidized to hemosiderin resulting in hemosiderinuria. Patients may have gross red-/tea-/cola-colored urine. Urine dipstick test is positive for blood but no RBCs are seen in microscopy. bPatients with chronic hemolysis have increased risk of calcium bilirubinate gallstones formation. Abbreviation: LDH, lactate dehydrogenase. | ||
Autoimmune Hemolytic Anemia
| ||
Direct Coomb’s test is positive with following reagenta | Anti-C3bb | |
| ||
aDirect and indirect Coomb’s (antiglobulin) agglutination test. bIgM antibodies are powerful activators of complement. In cold auto-immune hemolytic anemia, complement proteins are bound to RBCs. cOther immunosuppressive agents that can be used are azathioprine, cyclosporine, and cyclophosphamide. dIn IgG-type autoimmune condition, blood cell destruction takes place in spleen. eTreatment is similar to other IgG-mediated diseases (e.g., immune thrombocytopenia). Abbreviations: EBV, Epstein-Barr virus; IgG, immunoglobulin G; IgM, immunoglobulin M; IVIG, intravenous immunoglobulin; SLE, systemic lupus erythematosus. | ||
Hereditary Spherocytosis
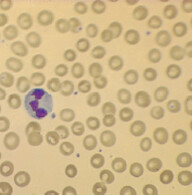
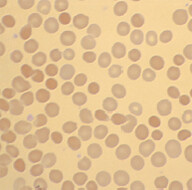
Background: It is mostly of autosomal dominant inheritance. It is due to genetic defect of structural proteins that are involved in giving the biconcave shape to RBCs. One of the commonly involved protein is called spectrin.
Pathophysiology: RBCs in this disorder are spherocytic, they cannot squeeze through the splenic sinusoids and are subsequently eaten up by splenic macrophages. These patients have chronically elevated bilirubin (due to chronic hemolysis), which in turn increases the risk of calcium bilirubinate gallbladder stones formation.
When to suspect: Features of extravascular hemolytic anemia with positive family hx, +/- biliary colic. Spherocytes are seen in peripheral blood smear and direct Coomb’s test is negative. Work-up: Best screening test is eosin-5-maleimide binding test (a flow cytometric test). If this is not available, other tests that can be done are osmotic fragility test, acidified glycerol lysis time test, or the cryohemolysis test.
Management: Oral folate replacement and transfusions as required. If moderate to severe anemia, splenectomy can be done (remove the organ where the RBCs are being destroyed).
How to differentiate between autoimmune IgG hemolytic anemia and hereditary spherocytosis?
Glucose-6-Phosphate Dehydrogenase Deficiency
Background: This is an X-linked recessive
hereditary disorder with deficiency of glu-cose-6-phosphate dehydrogenase (G6PD), which is a powerful antioxidant in RBCs. In patients of African American descent, it usually presents in a milder form, whereas patients of Mediterranean descent have more severe form.
Precipitating factors: Acute intravenous hemolysis occurs whenever there is an increased oxi-dative stress:
Any infection (MC precipitaing factor).
Drugs: sulfamethoxazole, primaquine, quinine, dapsone, isoniazid,
nitrofurantoin, etc.
14 Isoniazid + anemia: think of G6PD deficiency (when acute) or sideroblastic anemia (when presentation is insidious)
Thrombosis is the MCC of death in paroxysmal nocturnal hemoglobinuria (PNH).
Work-up:
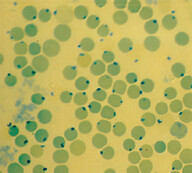
Rx: Supportive transfusion during an acute episode, treat underlying problem and avoid oxidant stress.
Paroxysmal Nocturnal Hemoglobinuria
Background: It is due to hereditary genetic defect in red cell membrane protein—phosphatidy-linositol glycan (PIG-A), aka decay-accelerating factor (proteins CD55 and CD59). This leads to easy binding of complement system to the red cell causing intravascular lysis of RBC, especially in acidic environment. This disorder is also associated with increased risk of varying degree of bone marrow failure (causing pancytopenia) and myelodysplastic syndrome.
Clinical pathophysiology: Nocturnal hemolysis can occur due to development of mild acidosis related to sleep-induced hypoventilation. Patients may present with early morning red urine. Hemolysis can also develop after exercise.
Along with acute hemolysis, patients also have increased risk of venous thrombosis, especially in uncommon locations such as hepatic vein or cerebral vein. Arterial thombosis has also been reported.
Work-up: NSIDx is flow cytometry, which will show absent or reduced expression of CD55/CD99. Older methods of detection are acidified hemolysis test (Ham’s test) and sugar water test (sugar is metabolized by RBCs into lactic acid).
Management
Supportive: Blood transfusion as needed and iron/folate supplementation.
For ongoing hemolysis, prednisone (steroids) might be helpful.
For patients with significant disease, give eculizumab (monoclonal antibody that inhibits complement activation).
In patients with severe disease, bone marrow transplantation can be done.
7.1.5 Sickle Cell Anemia
Background: It occurs due to single nucleotide (point) mutation in β-globin gene. This mutation replaces the amino acid glutamate (which is hydrophilic) with valine (which is hydrophobic) in the sixth position of β-globin protein. This change of a single amino acid increases risk of change in RBC shape, from biconcave to sickle shaped, whenever the hemoglobin is relatively desaturated. These sickled cells can clump together and easily get stuck in the vessels, obstructing the blood flow, thus causing infarction in various organs.
Diagnosis
For acute presentation, NSIDx is peripheral blood smear to look for sickled cells. To make the definitive diagnosis, use hemoglobin assay.
Sickle cell traita | |
a Most of the time, patients with sickle cell trait are asymptomatic or can have minor renal manifestations such as microscopic or macroscopic hematuria and/or isosthenuria. 16 Isosthenuria is loss of concentrating or diluting ability of kidney that results in urine having the same specific gravity as plasma. (Iso = same) | |
Acute presentation of sickle cell disease (acute anemia and acute painful crises)
7.1.6 Acute Anemia in Sickle Cell Disease
NSIDx is CBC, reticulocyte count and peripheral blood smear.
Peculiar PNH can cause venous and arterial thrombosis and is treated with eculizumab.
Rx: is supportive with RBC transfusion as needed and treatment of underlying cause.
Acute vaso-occlusive sickling crisis
Acute clumping of sickled RBCs in the arteries and arterioles can cause ischemia/infarction. Depending on location, it can present with the following:
Stepwise management of acute vaso-occlusive sickling crisis
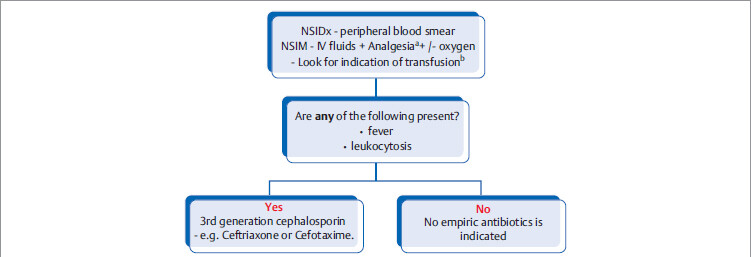
Chronic/Preventive management of patients with sickle cell disease
Within the first 3 months of age, prophylactic penicillin is initiated and continued until age 5. After this, decision to continue prophylaxis is made on a case-by-case basis.
All patients with underlying chronic hemolysis should be on oral folate therapy.
In symptomatic patients, hydroxyurea is recommended. Hydroxyurea increases production of HbF and decreases production of HbS, thus decreasing risk of sickling crisis.
Functional asplenia: By the time patients reach late childhood, they usually have fibrosed spleen due to recurrent splenic infarction (rightly called autosplenectomy). So, all patients with sickle cell disease should receive prophylaxis indicated in asplenia.
Vaccination and antibiotic prophylaxis for patients with splenectomy or functional asplenia
Vaccination: Spleen plays important role in immune defense against encapsulated microorganisms. Patients undergoing splenectomy, or with functional asplenia should be vaccinated against the following organisms:
In patients underoing splenectomy, administer vaccination either 2 weeks prior to surgery (preferred), or 2 weeks after the surgery | |
Asplenic children receive daily oral penicillin prophylaxis for at least 1 year or until age of 5, whichever is later. Elective splenectomy (e.g., for treatment of hereditary spherocytosis) is usually deferred until age 6, but if needed due to severe anemia, partial splenectomy can be done earlier.
Peripheral blood smear findings in asplenia:
5. Patient with hx of intermittent cola-colored urine presents with history of right upper quadrant (RUQ) pain usually after eating. What is the likely dx?
6. Patient develops abdominal pain and jaundice in the setting of 3-day history of fever and increased frequency of urination. CBC reveals acute anemia and elevated bilirubin. Urine analysis (UA) is positive for dipstick blood but no RBCs are seen microsopically. What are the differential dx?
7. African American man presents with hx of long-standing asymptomatic gross and microscopic hematuria. CT abdomen reveals no renal or urologic issues. What is the NSIDx?
In all chronic hemolytic anemias (i.e., sickle cell disease, thalassemias, chronic autoimmune hemolytic anemia, hereditary spherocytosis, etc.), if there is acute anemia with low reticulocyte count, think of the following:
Acute folate deficiency: all patients with chronic hemolysis should be on oral folate replacement.
Parvovirus B19 infection: can cause acute aplastic crisis (pancytopenia) or pure red cell aplasia (only anemia).
Work-up: If folate is normal, consider testing for parvovirus B19 IgM antibody and PCR.
7.1.7 Normocytic Anemia with Zero or Low Reticulocytic Count
In this case, bone marrow is not producing new RBCs. In anemic patients with low reticulocyte count and no obvious cause,
17 Normocytic anemia with reduced reticulocyte count can develop due to following conditions:
• Advanced chronic kidney disease leads to erythropoietin (epo) deficiency. (Epo stimulates marrow stem cells to produce RBCs.)
• Severe medical illness (e.g., severe alcoholism, endocrine disease, infection, etc.).
• Deficiency of raw materials needed to make RBCs (iron, folate, or vitamin B12 deficiency).
In these cases, if no other features of bone marrow disorder are present, bone marrow biopsy can be deferred.
Absent or reduced stem cells in the marrow which involves all cell lineage | |
Pure red cell aplasiaa | |
Overcrowding of bone marrow space with dysplastic or cancerous malignant cells | |
aThis is associated with parvovirus infection and thymoma. Congenital form of pure cell aplasia is Blackfan-Diamond syndrome (look for skeletal abnormalities). | |
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


