Thrombotic Thrombocytopenic Purpura, Hemolytic-Uremic Syndrome, and Related Disorders
Thrombotic Thrombocytopenic Purpura, Hemolytic-Uremic Syndrome, and Related Disorders
Han-Mou Tsai
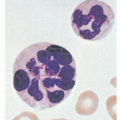
The various forms of thrombocytopenia discussed in this chapter share the common features of thrombocytopenia and hemolysis with characteristic schistocytes on blood smears (microangiopathic hemolytic anemia, MAHA) (Fig. 48.1). It is believed that thrombocytopenia results from consumption of platelets, whereas erythrocyte fragmentation and hemolysis are due to mechanical injury of the red blood cells by abnormal levels of shear stress.
Fragmentation of the red blood cells occurs in two types of clinical conditions: vascular devices such as prosthetic heart valves, ventricular assist devices, and extracorporeal oxygenator and microvascular stenosis.
In the absence of mechanical devices, fragmentation of the red blood cells signifies stenosis in the arteriolar microvasculature. This is because wall shear stress, determined by blood viscosity, flow rate, and the inverse of the luminal diameter to the third order, is at its highest in the arterioles and may be further increased to exceed the threshold level of red cell fragmentation when the lumen is narrowed. Furthermore, the red cells are likely to be entrapped in the presence of microvascular stenosis. The combination of abnormal shear stress and cell entrapment accounts for red cell fragmentation in patients with arteriolar stenosis. Due to their lower shear stress profile, it is unusual that stenosis in the venules, veins, or arteries is sufficient to cause red cell fragmentation.1
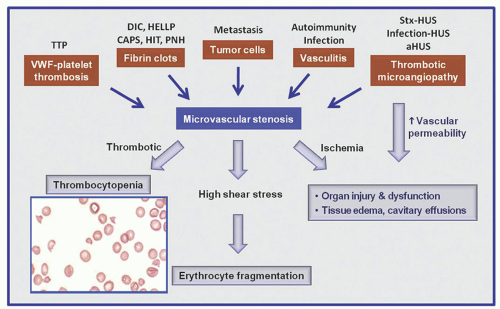
Arteriolar stenosis may result from one of five different types of pathology (Fig. 48.1): (1) von Willebrand factor (vWF)-platelet thrombosis, as typically observed in patients with thrombotic thrombocytopenic purpura (TTP) due to severe ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif, member 13) deficiency; (2) platelet fibrin thrombosis, as exemplified in patients with disseminated intravascular coagulopathy (DIC) but also may occur in a variety of other conditions; (3) tumor cell invasion of the microvasculature in patients with metastatic neoplasm; (4) microvascular vasculitis, as occasionally observed in patients with systemic lupus erythematosus and related autoimmune disorders or certain types of infections such as Rocky Mountain spotted fever and anthrax; and (5) thrombotic microangiopathy (TMA), as observed in patients with the typical shiga toxin-associated hemolytic-uremic syndrome (stx-HUS) following Escherichia coli infection, pneumococcal HUS, or atypical hemolytic-uremic syndrome (aHUS) due to defective regulation of the alternative complement pathway.
FIGURE 48.1. A scheme depicting how different types of pathology may lead to microvascular stenosis and the syndrome of thrombocytopenia and microangiopathic hemolysis (MAHA). Thrombotic microangiopathy may also lead to tissue edema and organ dysfunction by increasing vascular permeability. DIC, disseminated intravascular coagulopathy; CAPS, catastrophic antiphospholipid antibody syndrome; HELLP, hemolysis with elevated liver enzymes and low platelets; HIT, heparin-induced thrombocytopenia; HUS, hemolytic-uremic syndrome; PNH, paroxysmal nocturnal hemoglobinuria; Stx-HUS, shiga toxin-associated hemolytic-uremic syndrome; TTP, thrombotic thrombocytopenic purpura.
A comprehensive classification of microangiopathic disorders is listed in Table 48.1. This classification includes four groups: TTP, TMA due to defective complement regulation (aHUS), TMA due to other mechanisms, and MAHA due to other types of pathology. In TMA, organ dysfunction may result from thrombotic stenosis, which causes thrombocytopenia and MAHA; non-thrombotic stenosis, which causes MAHA but not thrombocytopenia; and/or abnormal vascular permeability, which causes tissue edema and cavitary effusions but not thrombocytopenia or MAHA. Due to multiple pathogenetic pathways, organ dysfunction or clinical disease severity in patients with aHUS or other types of TMA does not always closely correlate with thrombocytopenia or MAHA. TTP, stx-HUS, and aHUS are further discussed in this chapter.
TABLE 48.1 CLASSIFICATION OF MICROANGIOPATHIC DISORDERS
I. TTP: a disease with propensity to microvascular thrombosis due to ADAMTS13 deficiency
Acquired: autoimmune inhibitors of ADAMTS13
Hereditary: mutations of ADAMTS13
II. aHUS: a disease with propensity to TMA due to defective complement regulation
A: Idiopathic aHUS
–
Mutations or genetic variants of CHF, MCP, CFI, CFB, C3, THBD, etc.
–
Autoantibodies to CFH, with or without CFHR1 genomic deletion
B: Co-morbidity as a trigger of aHUS presentation in patients with the disease
–
Pregnancy, IV contrast, pancreatitis, infection, inflammation, surgery, trauma, etc.
Ventricular assist device, extracorporeal membrane oxygenator, prosthetic heart valves, etc.
ADAMTS13, a disintegrin and metalloprotease with thrombospondin type 1 motif, member 13; aHUS, atypical hemolytic-uremic syndrome; CAPS , catastrophic antiphospholipid antibody syndrome; CFB, complement factor B; CFH, complement factor H; CFHR1, complement factor H-related protein 1; CFI, complement factor I; DIC, disseminated intravascular coagulopathy; HELLP, hemolysis with elevated liver enzymes and low platelet count; HUS, hemolytic-uremic syndrome; HIT, heparin-induced thrombocytopenia; MCP, membrane cofactor protein (CD46); PNH, paroxysmal nocturnal hemoglobinuria; THBD, thrombomodulin; TMA, thrombotic microangiopathy; TTP, thrombotic thrombocytopenic purpura; vWF, von Willebrand factor.
*Some patients are found to have aHUS with defective complement regulation.
**Severe hypertension may be a consequence of aHUS rather than cause of thrombotic microangiopathy
ACQUIRED THROMBOTIC THROMBOCYTOPENIC PURPURA
The current definition of TTP is based on the demonstration of autoimmune inhibitor for acquired TTP and genetic mutation of the ADAMTS13 gene for hereditary TTP. By this definition, TTP is a disease that is prone to, but does not always have, microvascular thrombosis, thrombocytopenia, microangiopathic hemolysis, or organ dysfunction.
This definition is different from the conventional diagnosis of TTP as a clinical syndrome. It requires the demonstration of ADAMTS13 inhibitors or mutations and encompasses patients who are asymptomatic and patients who do not have thrombocytopenia or microangiopathic hemolysis.
The problem of conventional TTP diagnosis based solely on clinical features is most clearly illustrated by the vast difference in the prevalence of severe ADAMTS13 deficiency among the patients in the Canadian Apheresis Group trial on the efficacy of plasma therapy and the patients in the Oklahoma TTP registry. Severe ADAMTS13 deficiency was detectable in more than 76% of the patients in the Canadian Apheresis Group trial, yet in less than 15% of the patients in the Oklahoma registry.1, 2, 3 Indeed, many clinical series of ‘TTP’ or ‘TTP/HUS’ included patients with atypical HUS or other causes of microangiopathic hemolysis, resulting in confusion in the diagnosis and management of patients presenting with microangiopathic hemolysis and thrombocytopenia.
Pathology
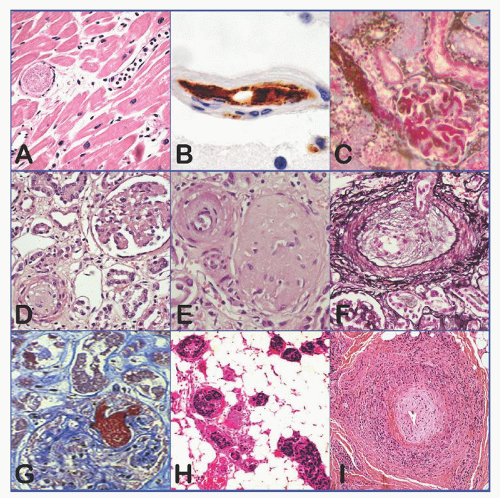
The pathology of TTP, as first described by Moschcowitz in 19254, is quite distinctive: widespread hyaline thrombi in the terminal arterioles and capillaries, accompanied by no or little endothelial injury or inflammation (i.e., microangiopathy) (Fig. 48.2A). The thrombi are present most extensively in the heart, pancreas, spleen, kidney, adrenal gland, and brain (mainly cerebral cortex) and are composed primarily of platelets and vWF (Fig. 48.2B).5, 6, 7 Small amounts of fibrin may be present surrounding or sometimes penetrating the amorphous or granular material. Glomerular microthrombi are usually spotty, and cortical necrosis of the kidney is uncommon in TTP. Fibrinoid necrosis and vascular or perivascular inflammatory cell infiltration are characteristically absent or minimal.
FIGURE 48.2. Histopathology and histochemistry of thrombotic thrombocytopenic purpura (TTP) and other microangiopathic disorders. A: TTP (heart); B: TTP (brain); C: shiga toxin-associated hemolytic-uremic syndrome (HUS) (kidney); D-F: atypical HUS (kidney); G: disseminated intravascular coagulopathy (DIC) (kidney); H: tumor cell invasion of microvasculature in metastatic neoplasm (soft tissue); I: proliferative vasculitis of lupus causing microvascular stenosis (soft tissue). The endothelial cells and the vessel wall are intact in TTP and DIC. All images are H&E stains except panel B (immunohistochemical stain for vWF, brown), panels C and G (fibrin stain of Carstair), and panel F (Jones silver stain).
In chronic cases, the thrombi may be infiltrated by fibroblasts or converted to subendothelial deposits by proliferating endothelial cell lesions. Pseudoaneurysmal dilatation may also be present upstream of the stenosis or occlusion.
In the literature, some investigators have suggested that endothelial cell injury is prominent in TTP. A review of the clinical features in those reports suggests that those patients likely had shiga toxin-associated or atypical HUS rather than TTP.
Pathophysiology
Historically two schemes have been proposed to account for the microvascular thrombosis in TTP: endothelial cell injury and uncontrolled platelet aggregation. The scheme of endothelial cell injury is not consistent with the absence or paucity of microangiopathy in TTP patients. The predominance of vWF and platelets in the thrombi and the integrity of vascular endothelial cells suggest that thrombosis results from dysregulation of vWF-platelet interaction in TTP.
vWF, a plasma glycoprotein derived primarily from vascular endothelial cells, supports platelet adhesion and aggregation at sites of microvascular injury under high shear stress conditions. Deficiency in vWF causes defective microvascular hemostasis and a bleeding diathesis in patients with von Willebrand disease.
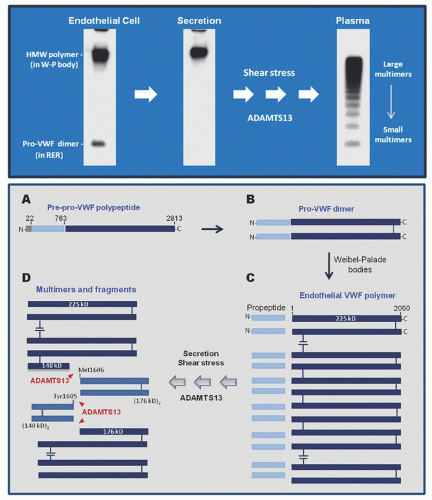
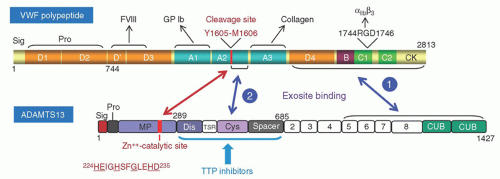
vWF is synthesized in vascular endothelial cells as a disulfide-bonded polymer that is converted to a series of multimers in the circulation (Fig. 48.3). This conversion is mediated by repetitive cleavage at the Tyr1605-Met1606 bond by ADAMTS13.
FIGURE 48.3. Gels and schemes depicting how VWF multimers are generated in the circulation. Upper panel: gels showing von Willebrand factor (vWF) polymer and multimers. In vascular endothelial cells vWF exists in two forms, a dimer of pro-vWF polypeptide in the rough endoplasmic reticulum (RER) and a polymer of mature vWF polypeptide in the Weibel-Palade bodies. Only the vWF polymer is secreted. Under the high shear stress conditions of the microcirculation, the vWF polymer is cleaved by ADAMTS13. A repetition of this process converts a vWF polymer to a series of multimers in normal plasma. vWF is separated by SDS-agarose gel electrophoresis and visualized with a radio-labeled antibody to vWF. Lower panel: a scheme of vWF biosynthesis and proteolysis. A: Pre-pro-vWF with 2,813 amino acid residues. B: A dimer of pro-vWF with disulfide bonds near the C-terminus. C: In the Weibel-Palade bodies, a vWF polymer is formed through disulfide bonding near the N-terminus of vWF dimers and cleavage of the propeptide. D: Under the high shear stress conditions of the microcirculation, vWF is cleaved at the Tyr1605-Met1606 bond, generating dimers of the 140 kD and 176 kD fragments and a series of progressively smaller multimers.
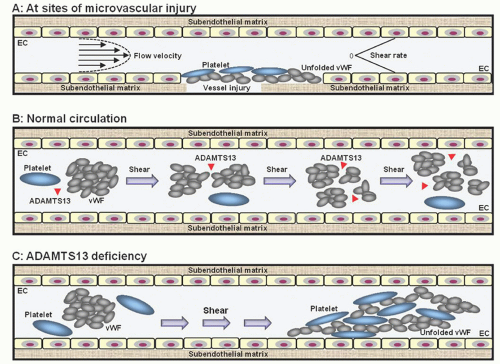
vWF exists in a compact form with its cleavage sites and platelet binding sites cryptic. This explains why in a test tube vWF is not susceptible to cleavage by ADAMTS13 and is inactive in aggregating platelets. High levels of shear stress induce a conformational change of vWF, exposing its binding sites for platelet receptors (Fig. 48.4A). Due to its large molecular size and responsiveness to shear stress, vWF is uniquely capable of supporting platelet adhesion and aggregation under high shear stress conditions.
In the circulation, the activation of vWF by shear stress is prevented by ADAMTS13, which cleaves vWF whenever its cleavage sites are exposed by shear stress. The process of proteolysis is repeated during each cycle of circulation through the microvasculature, thereby converting vWF from a large polymer to a series of multimers while maintaining vWF in its compact inactive configuration (Fig. 48.4B).
A disruption in this proteolytic regulation of vWF occurs in TTP, in which ADAMTS13 deficiency, due to genetic mutation or autoimmune inhibitors, leads to incessant unfolding and activation of vWF by shear stress, resulting in vWF-platelet binding and microvascular thrombosis (Fig. 48.4C). This explains why a severe deficiency of ADAMTS13 can lead to microvascular thrombosis characteristic of TTP.
FIGURE 48.4. A scheme depicting how ADAMTS13 deficiency leads to von Willebrand factor (vWF)-platelet aggregation and microvascular thrombosis seen in thrombotic thrombocytopenic purpura (TTP). A. The responsiveness of vWF to shear stress allows it to be activated at sites of microvascular injury to support platelet adhesion and aggregation. B. In the circulation, vWF-platelet aggregation is prevented by ADAMTS13, which cleaves vWF whenever its cleavage sites are exposed by shear stress. This process maintains vWF in its compact inactive configuration while its size becomes progressively smaller. C. In the absence of ADAMTS13, vWF is relentlessly activated by shear stress, leading to vWF-platelet aggregation and microvascular thrombosis of TTP. Thrombosis increases the shear stress in the microcirculation, leading to further cycles of vWF-platelet aggregation.
Observations in patients with TTP reveal that vWF-platelet aggregation and thrombosis does not occur when the plasma ADAMTS13 activity level is greater than 10% of normal. On the other hand, a patient with ADAMTS13 activity less than 10% may be asymptomatic and may have a normal platelet count. This is because other factors such as platelet number, vWF level, shear stress profile in the circulation, and modifiers of vWF response to shear stress such as thrombospondin8 may affect the propensity to vWF-platelet aggregation when ADAMTS13 is deficient.
Distinction between Thrombotic Thrombocytopenic Purpura Disease and Its Complications
A patient with autoimmune inhibitors or genetic mutations of ADAMTS13 may be completely asymptomatic and have normal blood counts. Such asymptomatic individuals are considered to have TTP, the disease, and are prone to the development of microvascular thrombosis causing clinical features such as thrombocytopenia, microangiopathic hemolysis, neurologic deficits, and injury of other organs.
Animal Models of ADAMTS13 Deficiency
Animal models of severe ADAMTS13 deficiency have been created in mice by inactivation of the ADAMTS13 gene and in baboons by infusion of an inhibitory ADAMTS13 antibody.9, 10, 11 In both models, thrombi comprising vWF and platelets ensue in the arterioles. The findings in these animal models support the role of ADAMTS13 deficiency in causing microvascular thrombosis of TTP. Nevertheless, some mouse strains are phenotypically free of thrombosis. The difference in phenotypic severity among the mouse strains highlights the principle of epistasis in which the composition of other genes modifies the expression of a molecularly defined disease.
Interactions between von Willebrand factor and ADAMTS13
The vWF polypeptide comprises a series of homologous domains. The cleavage site of vWF, Tyr1605-Met1606, is located in the A2 domain, sandwiched between the A1 domain, which contains epitopes interacting with the platelet glycoprotein Ib, and the A3 domain, where a collagen-binding epitope is located (Fig. 48.5). This topographic arrangement facilitates the regulation of platelet thrombus formation, as cleavage of vWF at the A2 domain disengages the platelet from the vessel wall.
Structural studies confirm that shear stress induces vWF cleavage by altering the conformation of vWF.12, 13 Narrow-angle neutron scattering studies of vWF in guanidine hydrochloride, which also promotes vWF cleavage by ADAMTS13,14 or after exposure to shear stress, shows that conformational change at the subdomain level is sufficient for proteolysis to occur.15, 16 Unfolding of vWF to extended configurations at the multimeric level is not necessary for proteolysis.
ADAMTS13, comprising 1,427 amino acid residues, is a member of the ADAMTS metalloprotease family, which shares a conserved domain structure of metalloprotease (MP)-disintegrin (Dis)-thrombospondin type 1 repeat (TSR)-cysteine rich region (Cys) and spacer (Spa) domains. ADAMTS13 contains 7 additional TSR downstream of the Spa domain, followed by two unique CUB (complement C1r/C1s, Uegf, Bmp1) domains (Fig. 48.5).
ADAMTS13 is synthesized primarily in the stellate cells of the liver.17, 18 ADAMTS13 may also be expressed, albeit at much lower levels by at least one to two orders, in the spleen and other organs. The localization of its biosynthesis to the stellate cells instead of hepatocytes may be instrumental in maintaining the ADAMTS13 activity in patients with hepatic insufficiency, as the stellate cells react to liver injury by activation and proliferation. The expression of ADAMTS13 in hepatic stellate cells and endothelial cells may be down-regulated by cytokines such as IFN-γ, TNF-α, and IL-4.19
FIGURE 48.5. A scheme depicting the domain structures of von Willebrand factor (vWF) and ADAMTS13 and their interactions. The cleavage of vWF by ADAMTS13 involves exosite and catalytic interactions. It is believed that the exosite binding 1 occurs constitutively, whereas exosite binding 2 occurs only when the sequence in the A2 domain downstream of the scissile bond Tyr1605-Met1606 is exposed by shear stress. The exosite bindings orient the catalytic site of ADAMTS13 to attack the scissile bond of vWF (double-arrowhead red line). The inhibitors of thrombotic thrombocytopenic purpura (TTP) patients target the exosites of ADAMTS13 downstream of the metalloprotease domain.
Expression of ADAMTS13 has been described in the renal glomerular podocytes and endothelial cells and vascular endothelial cells in culture.20, 21 The ADAMTS13 expressed in renal glomeruli may cleave vWF before it is neutralized by autoantibodies, accounting for the generally milder renal injury observed in acquired versus hereditary TTP.
The metalloprotease domain of ADAMTS13 contains a catalytic 224-HEIGHSFGLEHD-235 module characteristic of the ADAMTS proteases (conserved residues are underlined). Structural analysis has identified three surface exosites of ADAMTS13 in the Dis, Cys, and Spa domains that react with discrete epitopes of the vWF A2 sequence around and downstream of the Tyr1605-Met1606 scissile bond.22, 23
The exosite bindings greatly enhance the efficiency of catalytic reaction between ADAMTS13 and vWF. The vWF scissile bond and the exosite binding sites of the vWF A2 domain are normally cryptic, only exposed by tensile force to initiate interaction with ADAMTS13. Another exosite in the TSR5-CUB region of ADAMTS13 may interact with a constitutively exposed epitope in the N-terminal D4-CK region of vWF. This C-terminal binding may facilitate the interaction of the exosites upstream.
Alteration of von Willebrand factor Multimers in Thrombotic Thrombocytopenic Purpura
The involvement of vWF in the pathophysiology of TTP was first suggested by the intriguing observation that large multimers disappear during acute crisis of chronic relapsing TTP and yet ultra-large multimers are present during remission.24
The complexity of vWF multimers in TTP is due to two opposing processes acting upon plasma vWF in patients with ADAMTS13 deficiency: deficiency of ADAMTS13 leading to the presence of ultra-large multimers; and the activation of vWF by shear stress leading to the consumption and depletion of ultra-large and large multimers.
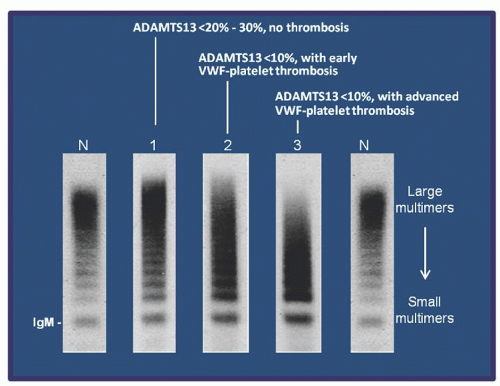
Three types of vWF multimer abnormalities are observed in TTP (Fig. 48.6). A shift of vWF to ultra-large forms occurs when ADAMTS13 activity is <20% to 30%; at this stage, no platelet thrombosis occurs (lane 1). The ultra-large and large multimers are partially depleted when ADAMTS13 is <10% and vWF-platelet aggregation begins to occur (lane 2). The ultra-large multimers are depleted and the large multimers are further decreased or depleted in patients presenting with extensive thrombosis and severe thrombocytopenia (lane 3).
The vWF changes in TTP are a dynamic process, as the multimers in a plasma sample represent a snapshot of the balance of the two changes acting in opposite directions. Transition from one multimer pattern to another occurs swiftly during the course of the disease, reflecting the changes in ADAMTS13 level and the process of vWF-platelet aggregation.
FIGURE 48.6. von Willebrand factor (vWF) abnormalities in thrombotic thrombocytopenic purpura (TTP) reflect the balance of underlying ADAMTS13 deficiency and vWF-platelet thrombosis. Three types of vWF abnormalities are detected in TTP during its course: Lane 1: A shift of vWF to ultra-large forms occurs when ADAMTS13 activity is <20%-30%. Lane 2: The ultra-large and large multimers are decreased when ADAMTS13 is <10%, and vWF-platelet aggregation begins to occur. Lane 3: The ultra-large multimers are depleted and the large multimers are further decreased in patients presenting with extensive thrombosis and severe thrombocytopenia.
In other types of microangiopathic hemolysis such as shiga toxin-associated or atypical HUS, depletion of the large vWF multimers also occurs. In these conditions, the loss of large multimers is due to excessive cleavage of vWF by ADAMTS13 under abnormal shear stress conditions.7
Other Pathogenic Mechanisms in Thrombotic Thrombocytopenic Purpura
Various hypotheses have been proposed to explain the thrombosis of TTP, including defects in fibrinolytic activity25 or prostacyclin homeostasis,26 increased circulating thrombomodulin, abnormal tissue plasminogen activator or plasminogen activator inhibitor-1 levels,27 antiendothelial cell antibodies,28, 29, 30 immune complexes,31 platelet aggregating proteins,32 anti-CD36 antibodies,33 calcium-dependent cysteine proteases,34 endothelial cell apoptotic factors,35 and activation of the complement system.36 These observations are preliminary, represent secondary changes, or are not specific for TTP.37, 38, 39, 40, 41 Immune complexes and activation of the complement system detected in patients with TTP may result from ADAMTS13 binding with its antibodies.
Causes of ADAMTS13 Deficiency
Severe ADAMTS13 deficiency results primarily from autoimmune inhibitors of the protease,42 although in a small number of patients the deficiency is the consequence of homozygous or, more commonly, compound heterozygous mutations of the ADAMTS13 gene.43
The ADAMTS13 activity level is also decreased in various pathologic conditions such as sepsis, malarial infection, multiorgan failure, major surgery, DIC, metastatic neoplasm, and pregnancy.44, 45, 46, 47, 48, 49, 50, 51 In these conditions, the decrease in ADAMTS13 levels is mild to moderate, and generally not sufficient to cause platelet aggregation. Yet the decrease induced by infection, surgery, or pregnancy may be sufficient to precipitate vWF-platelet aggregation and thrombosis in patients with pre-existing ADAMTS13 mutations or inhibitors, giving rise to the suspicion that these conditions cause TTP.
The ADAMTS13 activity is also less stable in pathologic samples, yielding falsely low ADAMTS13 levels if the samples are not properly handled.
Autoimmune Inhibitors of ADAMTS13
In patients with acquired TTP, which accounts for >95% of TTP cases, deficiency of ADAMTS13 results from autoimmune inhibitors of the ADAMTS13 protease.52 In most cases of acquired TTP the causes of the autoimmunity are unknown. It is speculated that an otherwise innocuous infection may induce autoimmune reaction to ADAMTS13 in genetically susceptible individuals. Indeed 10% to 40% of TTP patients exhibit positive autoimmune reactions to other antigens, suggesting they have defective immune regulation. Deranged immune regulation is consistent with the finding that the HLADRB1*11 allele is overrepresented among patients with acquired TTP.53
Ticlopidine therapy increases the risk of developing ADAMTS13 antibodies by 50- to 300-fold.54, 55 The antibodies occur between 2 to 8 weeks after institution of ticlopidine therapy, respond to plasma exchange and discontinuation of the culprit drug, and generally do not recur. Although another anti-platelet thienopyridine, clopidogrel, has also been implicated,56 an association between clopidogrel and TTP has not been confirmed.
HIV infection has been associated with TTP.57 It is estimated that the risk is increased by 35-fold among patients not being treated with antiretroviral therapy. This higher risk may be related to the predisposition of HIV-infected individuals to autoimmunity.
An ADAMTS13 polymorphism (R1060W) is reportedly more prevalent in acquired TTP patients than in the population, raising the speculation that certain ADAMTS13 polymorphisms may predispose the affected individuals to develop ADAMTS13 inhibitors.58 The validity of this association remains to be confirmed.
ADAMTS13 Mutations
DNA sequence analysis reveals that hereditary TTP patients have compound heterozygous or, less commonly, homozygous mutations of the ADAMTS13 gene on chromosome 9q34.59 More than 80 mutations, including nonsense, missense, frame-shifting insertion or deletion, and splicing mutations, have been detected in patients with hereditary TTP.
Only a few mutations have been detected in seemingly unrelated families. One mutation, 4143insA, has been detected in at least 15 patients in central-northern Europe, Turkey, and Australia that appear to share a common haplotype.60
More than 25 polymorphisms have also been detected in the coding sequence of ADAMTS13. One polymorphism, P475S, common among Japanese (5%), Koreans (4%), and Chinese (1%), makes the ADAMTS13 variant more susceptible to inhibition by urea,61, 62, 63 which is used in some ADAMTS13 assays.64 It yields falsely low ADAMTS13 activity levels if urea-based assays are used.65 Certain cis-combinations of polymorphisms or mutationpolymorphism may result in severe ADAMTS13 deficiency.66
Characteristics of the Inhibitors
In most patients the levels of the ADAMTS13 inhibitors are low (<10 U/ml), and often further decrease to undetectable levels after a few weeks or months. Occasionally the inhibitor level may surge to very high levels.
The ADAMTS13 inhibitors of TTP are primarily IgG, with IgA and IgM antibodies detectable infrequently.67 The VH1-69 germline heavy chain gene appears to be used most frequently in producing the ADAMTS13 antibodies.68 All four subclasses of IgG have been detected, although IgG4 appears to be the most prevalent, detectable in ≥90% of the patients according to one study.
Targets of the ADAMTS13 Inhibitors
The cysteine-rich region and spacer domain are an integral part of the epitopes of ADAMTS13 reacting with the inhibitors of TTP patients. ADAMTS13 variants truncated upstream of the cysteine-spacer domain, but not those truncated downstream, exhibit markedly decreased but detectable vWF cleaving activity that is not suppressible by the inhibitors of patients with TTP.69 This decreased activity is consistent with the existence of critical exosites in the deleted domains. Such nonsuppressible ADAMTS13 species may be exploited to overcome the therapeutic difficulty posed by ADAMTS13 inhibitors.
Further mapping studies show that residues R660, Y661, and Y665 in the spacer domain are critical for binding with vWF A2 domain sequences.70 They are also the target of TTP inhibitors. Substitution of residues Arg660, Tyr661, or Tyr665 in ADAMTS13 with Ala abolishes the binding targets of ADAMTS13 inhibitors. Intriguingly, two vWF variants with substitutions of these three residues exhibit increased vWF cleaving activity by several fold. Such “super” variants of ADAMTS13 may be potential candidates as a replacement therapy for patients with TTP, although immune responses to these foreign proteins may be a concern.71
Some studies found that TTP antibodies may also target other epitopes downstream of the spacer domain. However, it remains unclear whether these antibodies contribute to ADAMTS13 deficiency in TTP patients.72
Clinical Presentation
The incidence of TTP is estimated to be 2 to 15 cases per 1 × 106 person-years.3, 48, 49 The broad range of the observed incidence rates is likely due to differences in demographics, particularly the prevalence rate of HIV infection in the population.
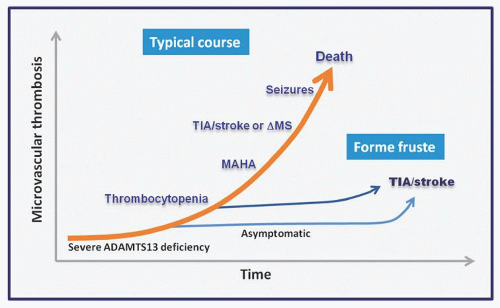
FIGURE 48.7. Progression of thrombotic complications in thrombotic thrombocytopenic purpura (TTP). When ADAMTS13 decreases below the threshold level, microvascular thrombosis begins to occur. In most patients, TTP evolves progressively from ADAMTS13 deficiency to thrombocytopenia, microangiopathic hemolytic anemia (MAHA), fleeting neurologic deficits (TIA), stroke, altered mental status, seizures, and death if it is not treated. Occasionally, a patient may be detected at the thrombocytopenia stage when a CBC is ordered for other reasons. TTP may also present in forme fruste, with microvascular thrombosis causing TIA or stroke before thrombocytopenia or MAHA occurs. ΔMS, altered mental status.
Acquired TTP primarily affects adolescents and adults, with median and mode ages between 30 and 40 years, although TTP may occasionally affect children <10 years of age. In most clinical series, the female-to-male ratio is 2 to 3:1.
Many patients present with the triad of thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and neurologic deficits or the triad with fever and renal dysfunction (pentad); neither is invariably present or specific for the diagnosis of TTP. In a typical case, the course of TTP begins with the appearance of ADAMTS13 inhibitors, which progressively decrease the plasma ADAMTS13 level, eventually leading to microvascular thrombosis, thrombocytopenia, microangiopathic hemolysis, and organ dysfunction (Fig. 48.7).
The duration of asymptomatic ADAMTS13 deficiency is quite variable, ranging from a few days to years. Serial monitoring of patients with a history of acquired TTP suggests that the plasma ADAMTS13 activity level may fluctuate for weeks to months before decreasing to less than 10% of normal and causing thrombosis.
Thrombocytopenia is detectable when platelet consumption exceeds compensatory platelet production. The stage of thrombocytopenia without overt hemolytic anemia may last for days to months, and the course of TTP at this stage is not invariably downhill; some patients may spontaneously revert to a normal platelet count. The patients at this stage may be given the incorrect diagnosis of immune thrombocytopenic purpura (ITP). On the other hand, infection, surgery, or pregnancy may worsen microvascular thrombosis in patients with subclinical ADAMTS13 deficiency, giving rise to the clinical impression that TTP is triggered by these events. Neurologic complications typically, albeit not invariably, present later, when thrombocytopenia is profound and microangiopathic hemolysis is evident.
Most of the patients have no other significant medical history and begin to notice ill-defined symptoms such as headache, dizziness, and fatigue that are often considered insignificant until more serious complications such as profound fatigue, focal neurologic deficits, syncope, mental status changes, or seizures ensue. Less frequently, a patient may present with abdominal pain, nausea, and vomiting, with or without pancreatitis, or chest pain due to myocardial infarction, or even sudden death.
Advanced renal failure causing oliguria, anuria, fluid retention, electrolyte abnormalities, or uremia is rare in acquired TTP. The creatinine generally increases only minimally. When advanced renal failure occurs in a patient with acquired TTP, it is likely due to other causes.
Thrombotic Thrombocytopenic Purpura without Microangiopathic Hemolysis or Thrombocytopenia
Thrombocytopenia and microangiopathic hemolysis are not invariably present at the time of acute presentation (Fig. 48.7). Relapse of TTP is also increasingly diagnosed before microangiopathic hemolysis ensues.
Until recently, TTP presenting with acute stroke or transient ischemic attack without overt microangiopathic hemolysis was diagnosed only in retrospect after the patients went on to develop thrombocytopenia and microangiopathic hemolysis. ADAMTS13 assays greatly facilitate the diagnosis of atypical TTP.
Hereditary Thrombotic Thrombocytopenic Purpura
Also known as Schulman-Upshaw syndrome or chronic relapsing TTP, hereditary TTP is rare, accounting for <5% of all TTP cases.73, 74 Because the diagnosis is not recognized in milder cases, the incidence is likely higher than presently appreciated.75
Hereditary TTP typically has its initial manifestations during the neonatal period, but historically is often not recognized until later in life.76, 77 Some of these patients have siblings who died before birth presumably due to TTP complications. Milder cases may present with thrombotic complications or have their disease recognized later in their life.76, 77, 78
In a typical case, the affected neonate is born with meconium stain or presents within a few hours after birth with neonatal distress, jaundice, and thrombocytopenia. Hemolysis may not be severe and schistocytes on blood smears may be overlooked. Occasionally, serious complications such as seizures and mental obtundation may occur.
The symptoms typically improve immediately after blood transfusion or exchange transfusion performed unknowingly for thrombocytopenia or hyperbilirubinemia. Consequently, the neonates may be discharged from the hospital without a correct diagnosis, only to present with complications of the disease weeks or years later.
Like most genetic disorders, hereditary TTP varies in its severity. Many patients with hereditary TTP invariably develop hematologic or other complications if not treated regularly with plasma infusion every 2 to 4 weeks. Others have no or only subtle symptoms and maintain normal or mildly subnormal platelet counts and develop more serious complications only intermittently. The severity may also vary during the lifetime of individual cases, with or without apparent exacerbating conditions such as pregnancy.
The thrombotic process may cause focal neurologic deficits, seizures, pancreatitis, or renal failure.79, 80 Of the patients not receiving regular plasma infusion, 10%-20% of the cases develop at least one episode of acute renal failure requiring dialysis during their course. The renal failure is reversible if the patients promptly receive plasma infusion. Chronic renal failure occurs in approximately 10% of the patients not being regularly treated with plasma infusion, likely a result of cumulative microinfarcts in the kidney.
The propensity to acute and chronic renal disease in hereditary TTP is quite different from its rarity in acquired TTP. It is speculated that local expression of ADAMTS13 in renal glomerular podocytes and endothelial cells20 may provide protection against vWF-platelet aggregation before its activity is suppressed by the inhibitors. Such protection is not present in patients with genetic ADAMTS13 mutations. Rarely renal failure may occur because the patient also has concurrent atypical HUS.81
Laboratory Findings
In de novo TTP cases presenting at the emergency service, the diagnosis is typically raised by the laboratory findings of severe thrombocytopenia and hemolysis with the presence of schistocytes on blood smears. The haptoglobin level is decreased or undetectable. Other common laboratory tests for assessment of hemolysis, including reticulocyte count, LDH, and indirect bilirubin are often but not invariably abnormal. In chronic cases, hemolytic anemia may predominate, as active platelet consumption is masked by compensatory thrombopoiesis.
As mentioned above, a patient with TTP may have no symptoms or may present with acute focal neurologic deficits without thrombocytopenia and/or microangiopathic hemolysis. Therefore, absence of thrombocytopenia or microangiopathic hemolysis in laboratory tests does not exclude the diagnosis of TTP.
Hematuria and proteinuria reflect renal glomerular thrombosis. The increase in the creatinine level is minimal in most cases. This is consistent with the pathologic findings that the kidney is minimally affected and its architecture is well preserved in TTP patients at autopsy.
Overt renal failure causing hypertension, fluid retention, electrolyte derangement, oliguria, or anuria favors the diagnosis of typical or atypical HUS over autoimmune TTP, unless there is a concurrent renal disorder. As mentioned above, acute and chronic renal failure may occur in patients with hereditary TTP.
Imaging studies such as CT or MRI are often requested for neurologic dysfunction. In most cases, the studies yield negative results or reveal only subtle ischemic changes, although occasionally a TTP patient may present with macrovascular ischemic strokes detectable by conventional imaging studies. It is speculated that in such cases the macrovascular thrombosis may result from thrombotic injury of the vasa vasorum.
The literature includes reports of “TTP” case series with a high percentage showing abnormal CT or MRI findings. However, a review of cases in these reports reveals that most of these patients likely had typical or atypical HUS rather than TTP.
Abnormal electrocardiograms with nonspecific ST-T wave changes and elevated CPK or troponin levels are common. Occasionally thrombosis may affect large coronary arteries leading to myocardial infarction. Cardiac arrhythmia is uncommon. Electromechanical dissociation, heart failure, or pulmonary infiltrates or hemorrhage occur in advanced, pre-terminal cases.
The amylase and lipase levels may be elevated. Abnormal liver function is rare. Abdominal CT scans may detect focal pancreatitis, intestinal wall or mesenteric thickening, or stranding due to ischemia or infarction.
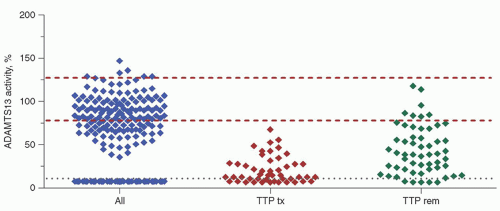
FIGURE 48.8. Plasma ADAMTS13 activity is undetectable in patients with thrombotic thrombocytopenic purpura (TTP). Patients (338 cases) presenting with thrombocytopenia and microangiopathic hemolysis segregate in two nonoverlapping groups based on their plasma ADAMTS13 activity levels. Severe ADAMTS13 deficiency defines the diagnosis of TTP in 219 patients (65%), of which 191 had autoimmune ADAMTS13 inhibitors and 28 had genetic mutation as the cause of the deficiency. ADAMTS13 activity is detectable in some patients after receiving plasma or blood transfusion (TTP tx). During remission, (TTP rem) most patients continue to have decreased ADAMTS13 activity levels, consistent with the chronic nature of their disease. The two upper dashed lines encompass the normal range of plasma ADAMTS13 activity. The lowest line marks the detection limit of the assay at 10%.
ADAMTS13 Activity Levels
Among patients presenting with thrombocytopenia and microangiopathic hemolysis, only TTP patients have severe ADAMTS13 deficiency (less than 10%), whereas patients with other disorders have detectable ADAMTS13 levels (Fig. 48.8). ADAMTS13 may be detectable if the assay is performed after the patients receive plasma or blood transfusions. The ADAMTS13 activity level remains decreased in most patients during their remission, suggesting that the autoimmunity is active. Severe ADAMTS13 deficiency provides the diagnosis of TTP in the patients with co-morbidities in whom the diagnosis would otherwise be difficult to make.
The assay used to obtain the data presented in Figure 48.8 uses purified vWF as the substrate and SDS-PAGE to detect the proteolytic products. This assay is technically demanding and not practical for use in clinical laboratories. Various assays using vWF peptides spanning the scissile bond have been developed for analysis of ADAMTS13 activity in clinical laboratories. The assays differ in their performance and reliability and may not provide a definitive diagnosis or exclusion of TTP.82, 83, 84, 85, 86, 87, 88
Because some clinical assays may yield falsely high or low values of ADAMTS13, the diagnosis of TTP requires corroboration of the ADAMTS13 test results with clinical and other laboratory data.
ADAMTS13 Inhibitors
Inhibitors of ADAMTS13, measured by mixing patient and normal plasma samples, are detectable in 80% to 90% of the acquired TTP patients. A negative test for ADAMTS13 inhibitors does not exclude the presence of ADAMTS13 inhibitors. In such patients, the presence of inhibitors may be inferred if the increase of the ADAMTS13 activity level is less than expected after plasma therapy, or if the ADAMTS13 activity recovered to >10% during remission.
ADAMTS13 Antibody and Antigen Assays
Assays have been developed to measure ADAMTS13 antigen and antibody levels in enzyme-linked assay formats. These solid-phase microtiter plate-based assays have limited utility in practice. The antigenic assay detects free ADAMTS13 as well as ADAMTS13/inhibitor complex, yielding low ADAMTS13 antigen levels in <50% of TTP patients.89 The ADAMTS13 antibody assay is highly sensitive (>90%) for acquired TTP but may yield false-positive results in 5% to 10% of individuals without ADAMTS13 antibodies.90, 91
Only gold members can continue reading. Log In or Register to continue