INTRODUCTION
SUMMARY
The human erythrocyte membrane consists of a lipid bilayer containing transmembrane proteins and an underlying membrane skeleton, which is attached to the bilayer by linker protein complexes. The membrane is critical in maintaining the unique biconcave disk shape of the erythrocyte and enabling it to withstand the circulatory shear stress. The integrity of the membrane is ensured by vertical interactions between the skeleton and the transmembrane proteins, as well as by horizontal interactions between skeletal proteins. Inherited defects of membrane proteins compromise these interactions and alter the shape and deformability of the cells, which ultimately results in their premature destruction and hemolytic anemia. The disorders are typically autosomal dominant and exhibit significant clinical, laboratory, biochemical, and genetic heterogeneity.
Hereditary spherocytosis is a common condition characterized by spherically shaped erythrocytes on the blood film, reticulocytosis, and splenomegaly. The underlying defect is a deficiency of one of the membrane proteins, including ankyrin, band 3, α-spectrin, β-spectrin, or protein 4.2. This weakens the vertical membrane interactions, resulting in loss of membrane and surface area. Spherocytes have diminished deformability, which predisposes them to entrapment and destruction in the spleen. Hereditary elliptocytosis is characterized by the presence of elliptical erythrocytes on the blood film. The principal abnormality affects horizontal membrane protein interactions and typically involves α-spectrin, β-spectrin, protein 4.1R, or glycophorin C. The membrane skeleton is destabilized and unable to maintain the biconcave disk shape, which manifests as an elliptical distortion of the cells in the circulation. Hereditary pyropoikilocytosis is a rare, severe hemolytic anemia characterized by markedly abnormal erythrocyte morphology caused by defective spectrin. Southeast Asian ovalocytosis is largely asymptomatic and is caused by a defect in band 3. The blood film shows large oval red cells with a transverse ridge across the central area. Acanthocytosis is typified by contracted, dense erythrocytes with irregular projections, which may be seen in patients with severe liver disease, abetalipoproteinemia, various neurologic disorders, certain aberrant red cell antigens, and postsplenectomy. Stomatocytosis is a rare group of inherited disorders associated with abnormal membrane permeability and red cell cation content, which either cause overhydration or dehydration of the cells.
Acronyms and Abbreviations:
AE1, anion exchanger-1; αLELY, α-spectrin low-expression Lyon; αLEPRA, α-spectrin low-expression Prague; AGLT, acidified glycerol lysis test; ANK, ankyrin; AQP1, aquaporin-1; BCSH, British Committee for Standards in Haematology; BPG, 2,3-bisphosphoglycerate; CDAII, congenital dyserythropoietic anemia type II; EMA, eosin 5′-maleimide; 4.1R, erythrocyte isoform of protein 4.1; GLT, glycerol lysis test; GLUT-1, glucose transporter-1; GP, glycophorin; GP-A, -B, -C, -D, -E, various members of glycophorin family; GSSG, oxidized glutathione; HARP, hypobetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration syndrome; HE, hereditary elliptocytosis; HPP, hereditary pyropoikilocytosis; HS, hereditary spherocytosis; HSt, hereditary stomatocytosis; MAGUK, membrane-associated guanylate kinase; MARCKS, myristoylated alanine-rich C kinase substrate; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; OF, osmotic fragility; PKAN, pantothenate kinase-associated neurodegeneration; RhAG, Rh-associated glycoprotein; SAO, southeast Asian ovalocytosis; SDS-PAGE, sodium dodecylsulfate polyacrylamide gel electrophoresis; UGT1, uridine diphosphate glucuronosyltransferase 1.
The erythrocyte membrane plays a critical role in the function and structure of the red cell. It is a key determinant of the unique biconcave disk shape and provides the cell with a finely tuned combination of flexibility and durability. These properties enable the erythrocyte to withstand the circulatory shear pressure and allow it to undergo extensive and repeated distortion while negotiating the microvasculature and the spleen, thus ensuring survival during its average 120-day life span. The red cell membrane maintains a nonreactive surface so that erythrocytes do not adhere to the endothelium or aggregate and occlude capillaries. It provides a barrier with selective permeability, which retains vital components inside the cell and permits the efflux of metabolic waste. To facilitate the transfer of carbon dioxide and to maintain pH homeostasis, the membrane exchanges chloride and bicarbonate anions, and it also actively controls the cation and water content of the erythrocyte. The membrane sequesters reducing agents required to prevent oxidative damage to hemoglobin and other cellular components, and it plays a role in regulating metabolism by reversibly binding and inactivating selected glycolytic enzymes.
Abnormalities of the erythrocyte membrane alter the shape of the cell and compromise its integrity and ability to survive the rigors of circulation, which leads to premature destruction and hemolysis. Erythrocyte membrane disorders comprise an important group of hereditary hemolytic anemias, which are classified according to the altered red cell morphology and include hereditary spherocytosis (HS), hereditary elliptocytosis (HE) and related disorders, and the hereditary stomatocytosis (HSt) syndromes. This chapter summarizes our current understanding of the erythrocyte membrane in normal cells followed by a discussion of the underlying molecular defects and their role in the pathophysiology and clinical manifestations of these disorders. The main emphasis is on spherocytosis and elliptocytosis, the two most common and best characterized diseases.
OVERVIEW OF THE ERYTHROCYTE MEMBRANE
The erythrocyte membrane is the most studied plasma membrane and serves as a paradigm for all cellular membranes. Mature erythrocytes are readily accessible; they contain no intracellular organelles, which facilitates the isolation of pure erythrocyte membranes; and “experiments of nature” resulting in abnormal erythrocyte morphology have provided unique opportunities to investigate the function of membrane components. These studies have revealed the primary structure and several important functions of the red cell membrane. Ongoing research, using the latest molecular technologies, continues to yield important insights into our understanding of membrane structure–function relationships, as well as genotype–phenotype correlations.
The erythrocyte membrane is a complex structure consisting of a relatively fluid lipid bilayer stabilized by an underlying two-dimensional membrane skeleton, which maintains the integrity of the biconcave disk shape of the erythrocyte (Fig. 46–1). The skeleton provides the cell with the strength and flexibility to deform rapidly and repeatedly and thus endure the shear stress encountered in the tiny capillaries of the microcirculation and in the spleen. The lipid bilayer separates the erythrocyte cytoplasm from the external plasma environment and contains phospholipids and cholesterol, as well as integral transmembrane proteins, which are tethered to the skeleton by interactions with linker proteins.
Figure 46–1.
Schematic model of the human erythrocyte membrane. The molecular assembly of the major proteins is indicated. Vertical interactions are perpendicular to the plane of the membrane and are represented by the ankyrin and junctional protein complexes that connect the membrane spectrin skeleton to the integral proteins embedded in the lipid bilayer. Horizontal interactions occur parallel to the plane of the membrane and involve spectrin tetramers and protein 4.1R. The proteins and lipids are not drawn to scale. b3, Band 3; GPA/GPC, glycophorin A/C; GLUT-1, glucose transporter-1.
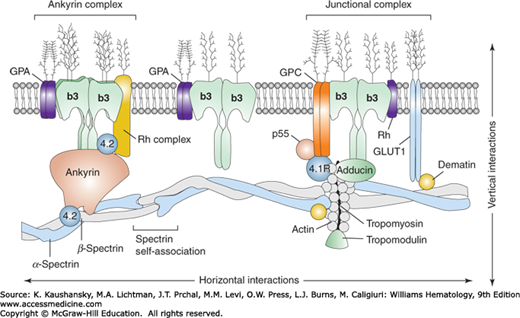
COMPONENTS OF THE ERYTHROCYTE MEMBRANE
The lipid bilayer comprises approximately 50 percent of the membrane mass and contains unesterified cholesterol and phospholipids in approximately equal amounts, with small amounts of glycolipids and phosphoinositides (Chap. 31).1,2 Mature erythrocytes are unable to synthesize fatty acids, phospholipids, or cholesterol de novo, and they depend on lipid exchange and limited phospholipid repair.3
Cholesterol regulates the fluidity of the membrane and is present in both leaflets, whereas the phospholipids are asymmetrically distributed. The choline phospholipids, phosphatidylcholine and sphingomyelin, are predominantly located in the outer leaflet and play a role in plasma lipid exchange and renewal of membrane phospholipids. Glycolipids carry several important red cell antigens, including A, B, H, and P, and are only found in the external leaflet with their carbohydrate moieties extending into the plasma. The aminophospholipids, phosphatidylserine and phosphatidylethanolamine, as well as phosphatidylinositol are located in the inner leaflet of the lipid bilayer.
This asymmetric distribution of phospholipids is maintained by a dynamic process involving flippase and floppase enzymes, which translocate the aminophospholipids to the inner and outer leaflets, respectively.4,5 A scramblase mediates bidirectional movement of phospholipids down their concentration gradient.6 Asymmetry of the phospholipids is important for the survival of the erythrocyte since exposure of phosphatidylserine on the outside surface of the cell, as found in sickle cell disease and thalassemia, has several deleterious consequences. It activates the coagulation cascade and may contribute to thromboses4; it facilitates adhesion to the vascular endothelium; it provides a recognition signal for macrophages to phagocytose these cells; and it decreases the interaction of skeletal proteins with the bilayer, which destabilizes the membrane.
Lipid rafts have been identified in erythrocytes.7 They form detergent-resistant membrane microdomains, enriched in cholesterol and sphingolipids, and are associated with several proteins, including stomatin and flotillin-1 and -2. These rafts play a role in signaling and invasion of malaria parasites.8
Pioneering studies resolved the major proteins of the red cell membrane by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and numbers from 1 to 8 were assigned to each protein starting with the largest protein, which migrated the slowest (Chap. 31).9 Subsequent research revealed minor bands between the major proteins and these were designated with decimals. Analysis of the individual proteins led to the renaming of some of them, such as band 1 and 2, which are now known as α– and β-spectrin, respectively. Technologic advances have enabled an in-depth analysis of the erythrocyte proteome by mass spectrometry, revealing a total of 340 membrane proteins.10 Table 46–1 summarizes the properties of the major components.
| Band | Protein | Mr (gel) | Mr (calc) | Copies per Cell (×103) | Percentage of Totala | Gene Symbol | Chromosomal Localization | Amino Acids | Gene Size (kb) | No. of Exons | Involvement in Hemolytic Anemias |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | α-Spectrin | 240 | 280 | 240 | 16 | SPTA1 | 1q22–q23 | 2429 | 80 | 52 | HE, HS, HPP |
| 2 | β-Spectrin | 220 | 246 | 240 | 14 | SPTB | 14q23–q24.2 | 2137 | >100 | 32 | HE, HS, HPP |
| 2.1 | Ankyrinb | 210 | 206 | 120 | 4.5 | ANK1 | 8p11.2 | 1881 | >100 | 40 | HS |
| 2.9 | α-Adducinc | 103 | 81 | 30 | 2 | ADDA | 4p16.3 | 737 | 85 | 16 | N |
| 2.9 | β-Adducinc | 97 | 80 | 30 | 2 | ADDB | 2p13–2p14 | 726 | ~100 | 17 | N |
| 3 | Anion exchanger-1 | 90–100 | 102 | 1200 | 27 | EPB3 | 17q21–qter | 911 | 17 | 20 | HS, SAO, HAc |
| 4.1 | Protein 4.1 | 80 | 66 | 200 | 5 | EL11 | 1p33–p34.2 | 588d | >100 | 23 | HE |
| 4.2 | Protein 4.2 | 72 | 77 | 200 | 5 | EB42 | 15q15–q21 | 691 | 20 | 13 | HS |
| 4.9 | Dematine | 48 + 52 | 43 | 40f | 1 | EPB49 | 8p21.1 | 383 | – | – | N |
| 4.9 | p55e | 55 | 53 | 80 | – | MPP1 | Xq28 | 466 | – | – | N |
| 5 | β-Actin | 43 | 42 | 400–500 | 5.5 | ACTB | 7pter–q22 | 375 | >4 | 6 | N |
| 5 | Tropomodulin | 43 | 41 | 30 | – | TMOD | 9q22 | 359 | – | – | N |
| 6 | G3PDg | 35 | 37 | 500 | 3.5g | GAPD | 12p13.31–p13.1 | 335 | 5 | 9 | N |
| 7 | Stomatin | 31 | 32 | – | 2.5 | EPB72 | 9q33–q34 | 288 | 12 | 7 | HSt |
| 7 | Tropomyosin | 27 + 29 | 28 | 80 | 1 | TPM3 | 1q31 | 239 | – | – | N |
| PAS-1 | Glycophorin Ah | 36 | – | 500–1000 | 85 | GYPA | 4q28–q31 | 131 | >40 | 7 | HE |
| PAS-2 | Glycophorin Ch | 32 | 14 | 50–100 | 4 | GYPC | 2q14–q21 | 128 | 14 | 4 | HE |
| PAS-3 | Glycophorin Bh | 20 | – | 100–300 | 10 | GYPB | 4q28–q31 | 72 | >30 | 5 | N |
| Glycophorin Dh | 23 | – | 20 | 1 | GYPD | 2q14–q21 | 107 | 14 | 4 | N | |
| Glycophorin E | – | – | – | – | GYPE | 4q28–q31 | 59 | >30 | 4 | N |
The membrane proteins are classified as either integral or peripheral based on the ease with which they can be removed from whole red cell membrane preparations in the laboratory. Integral or transmembrane proteins are embedded in the lipid bilayer by hydrophobic interactions and require detergents to extract them. They often protrude from the bilayer and extend into the plasma and/or the interior of the erythrocyte and these structural features correlate with their functions as transport proteins, receptors, signaling molecules, and carriers of red cell antigens.
Peripheral proteins constitute the membrane skeleton and are loosely attached to the cytoplasmic face of the lipid bilayer and can be extracted by high or low salt concentrations or by high pH. Attachment is mediated indirectly by covalent or noncovalent interactions with the cytoplasmic domains of the transmembrane proteins, as well as by direct interactions with the inner leaflet of the lipid bilayer. These associations are dynamic and the affinity of binding is regulated by post-translational modifications of the proteins, including phosphorylation, methylation, glycosylation, or lipid modification (myristoylation, palmitoylation, or farnesylation). Peripheral proteins typically function either as structural proteins and form part of the membrane skeleton or they serve as linker proteins attaching the skeleton to the bilayer.
Many erythrocyte proteins belong to superfamilies and have homologues in nonerythroid cells that are structurally related but are encoded by different genes. This genetic diversity explains why the clinical expression of most (but not all) red cell membrane protein mutations is confined to the erythroid lineage. Several proteins exist in different isoforms, created by tissue- and developmental stage-specific alternative splicing or by the use of alternative initiation codons or promoters. Many of the membrane proteins are large, multifunctional proteins and therefore the position of a mutation determines the functional abnormality and clinical phenotype.
The most abundant and important transmembrane proteins are band 3, which is the anion exchanger (AE1) of the erythrocyte, and the glycophorins (GPs).
The red cell contains approximately 1.2 million copies of AE1, a multifunctional and major integral membrane protein (see Table 46–1). It has a molecular mass of 102 kDa, but migrates as a diffuse band on sodium dodecylsulfate (SDS) gels as a result of heterogeneous N-glycosylation. The 911-amino-acid protein consists of two functional domains; an N-terminal 43-kDa cytoplasmic domain and a 52-kDa transmembrane channel, including a short 33-amino-acid C-terminal cytoplasmic tail11 (Fig. 46–2). The anion exchange domain encompasses 13 α helical transmembrane segments and one nonhelical segment all connected by hydrophilic loops.12,13 The short cytoplasmic tail binds carbonic anhydrase II to form a metabolon with the transmembrane domain, enabling the exchange of HCO3− and Cl− anions, which is a critical function of the red cell.14 The extracellular surface of the transmembrane domain of band 3 carries several antigens, including Diego, I/i, and Wright blood groups.
Figure 46–2.
Schematic model of human erythrocyte band 3. The N and C terminal regions of the protein extend into the cytoplasm and provide binding sites for several red cell proteins and enzymes. The transmembrane domain forms an anion exchange channel and consists of 13 α helical segments embedded in the lipid bilayer and one nonhelical segment. Asparagine 642 is linked to complex carbohydrates, which protrude on the exterior of the red cell. Tyrosine 8 is phosphorylated. The domains are not drawn to scale.
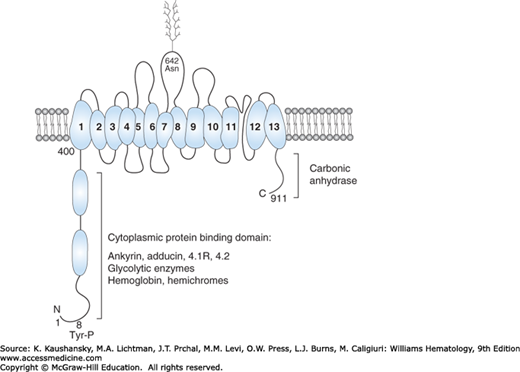
The N-terminal phosphorylated cytoplasmic domain serves as a major hub for protein-protein interactions, which perform key functions (see Figs. 46–1 and 46–2).15 It regulates metabolic pathways by sequestering key glycolytic enzymes such as glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase and aldolase, which are inactive when bound. Phosphorylation at tyrosine 8 prevents binding, which liberates the active enzymes.16 The cytoplasmic domain interacts with hemoglobin and hemichromes and plays a role in red cell aging17; it associates with several peripheral membrane proteins, including the erythrocyte isoform of protein (4.1R),18,19 protein 4.2,20 and adducin,21 as well as phosphatases and kinases. This domain also serves as the major attachment site of the membrane to the underlying skeleton through its interaction with ankyrin, which binds to spectrin (see Figs. 46–1 and 46–2).22,23
Band 3 associates with other transmembrane proteins to form macromolecular complexes (see Fig. 46–1).24 This includes the major GP, GPA,25 and the Rh protein complex, consisting of Rh-associated glycoprotein (RhAG), Rh, CD47, LW, and GPB (Chap. 136).24 In addition, band 3 participates in the protein 4.1-based junctional complex of proteins.19
Band 3 is encoded by the SLC4A1 gene, which produces different tissue-specific isoforms.11,26 The erythroid isoform is controlled by a promoter upstream of exon 1, whereas transcription of the kidney isoform is initiated from a promoter in intron 3, resulting in a protein lacking the first N-terminal 65 amino acids.
GPs are integral membrane glycoproteins composed of an extracellular hydrophilic N-terminal domain, a single α-helical membrane-spanning domain, and a C-terminal cytoplasmic tail. GPA, GPB, and GPE are homologous and are encoded by closely linked genes that arose by duplication of the ancestral GPA gene.27 GPC and GPD are encoded by the same gene but make use of alternate initiation codons.28
GPs have very high sialic acid content and are responsible for most of the external negative charge of red cells, which prevents the adherence of cells to each other and the vascular endothelium. The GPs carry a large number of blood group antigens, including MN, SsU, Miltenberger, En(a–), MK, and Gerbich (Chap. 136). They also function as receptors for Plasmodium falciparum, the most virulent malaria parasite. Within the lipid bilayer of the membrane, GPA interacts with band 3 as part of a macromolecular complex, and may serve as a chaperone for band 3 targeting to the membrane.25 GPC associates with protein 4.1R and p55, thereby providing an additional contact site between the membrane and the skeleton (see Fig. 46–1).19 These interactions play a role in stabilizing the membrane.
The Rh-RhAG group of proteins is part of a macromolecular band 3 complex, which stabilizes the membrane. RhAG belongs to the ammonium transporter family of proteins, but its function is controversial. Numerous other proteins are embedded in the lipid bilayer, many of which are implicated in clinical immunohematology and membrane disorders, such as the XK, Kell, Kidd, Duffy, and Lutheran glycoproteins (see Fig. 46–1).11,19 Additional integral membrane proteins include ion pumps and channels, such as stomatin, aquaporin, glucose transporter (GLUT-1), and various cation and anion transporters.
Underlying the lipid bilayer is the peripheral membrane skeleton, an interlocking network of structural proteins, which plays a critical role in maintaining the shape and integrity of the red cell. The major proteins of the erythrocyte membrane skeleton are spectrin, actin, proteins 4.1R, 4.2, 4.9, p55, and the adducins, which interact in a horizontal plane. Linker proteins mediate the vertical attachment of the skeleton to integral membrane proteins in the lipid bilayer (see Fig. 46–1). The primary connecting protein is ankyrin, which links spectrin to the cytoplasmic domain of band 3, as well as to the Rh–RhAG complex. Protein 4.1R provides an additional link with GPC and band 3.
Spectrin is the major constituent of the erythrocyte membrane skeleton and is present at approximately 240,000 molecules per cell.29 It is a multifunctional protein composed of two homologous but structurally distinct subunits, α and β, encoded by separate genes, which may have evolved from duplication of a single ancestral gene30 (see Table 46–1 and Fig. 46–3). Both α– and β-spectrin contain tandem homologous spectrin repeats that are approximately 106 amino acids long and are folded into three antiparallel helices, A, B, and C. Each repeat is connected to the adjacent repeat by short ordered α-helical linkers (Fig. 46–3).31,32 Erythrocyte α-spectrin is a 280-kDa protein comprising 20 complete repeats, an N-terminal partial repeat, a central SH3 domain, and a C-terminal calcium-binding EF hand. The β-spectrin subunit is a 246-kDa polypeptide consisting of 16 complete repeats, an N-terminal actin binding domain, a partial repeat near the C-terminus, and a nonhomologous phosphorylated C-terminus. Mild trypsin treatment of spectrin cleaves the two subunits into distinct structural domains: αI-V and βI-IV. The triple helical structure of the spectrin repeats renders the molecule highly flexible and enables it to extend and condense reversibly, which provides the red cell with elasticity and durability to withstand the shear stress encountered in the circulation.
Figure 46–3.
Schematic model of human erythrocyte α– and β-spectrin. The proteins consist of multiple homologous spectrin repeats of approximately 106 amino acids numbered from the N-terminal. Each repeat is composed of three α helices. Nonhomologous regions include an SH3 domain and calcium-binding EF hands in α-spectrin, a protein 4.1R binding domain, and a C-terminal phosphorylated tail in β-spectrin. The nucleation site indicates the initial region of interaction between α and β monomers to form an antiparallel heterodimer. Spectrin heterodimer self association into tetramers involves helix C of the α0 partial repeat of α-spectrin and helices A and B of the partial β17 repeat of β-spectrin to form a complete triple helical repeat. Ankyrin binds to repeats 14 and 15 of β-spectrin. Limited tryptic digestion of spectrin cleaves the proteins into discrete αI-V and βI-IV domains.
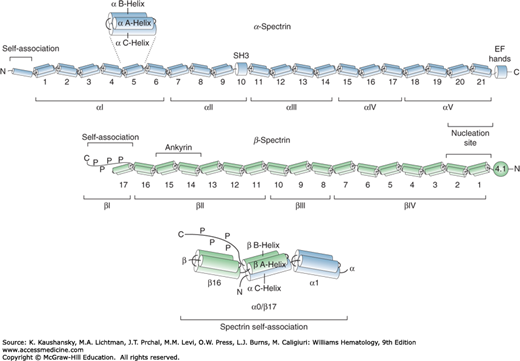
The core structure of the erythrocyte skeleton consists of spectrin heterotetramers, which are strong but flexible filaments. Tetramers are assembled from the monomers in a series of events. For initial heterodimer formation, the α– and β-spectrin chains align in an antiparallel fashion and interact with high affinity through long-range electrostatic interactions at a nucleation site, comprising repeats α20–21 and β1–2.33 This triggers the association of the remaining repeats in the two subunits in a zipper-like fashion. Repeats at the N-terminus of α-spectrin (αI domain) and the C-terminus of β-spectrin (βI domain) are the regions involved in heterodimer self-association to form tetramers. Partial repeat β17 consists of two helices (A and B) which interact with the single helix C of partial repeat α0 to form a complete triple helical repeat (see Fig. 46–3). The interface of this tetramerization site is dominated by hydrophobic contacts supplemented by electrostatic interactions.34 Phosphorylation of the C-terminal region of β-spectrin beyond the self-association site decreases the mechanical stability of the membrane.
At the opposite tail end of the spectrin tetramers, the N terminus of β-spectrin binds to short F-actin filaments, which is potentiated by 4.1R, to form the core of a junctional complex,35 which links six tetramers together into a hexagonal skeletal network (Fig. 46–4).36 The C-terminal EF hand of α-spectrin enhances this spectrin-actin-4.1R interaction.37 Numerous other proteins participate in the junctional complex, including adducin, protein 4.9, p55, tropomodulin and tropomyosin (see Fig. 46–1).19 Protein 4.1R binds to GPC and band 3, which serves as a secondary attachment site of the skeleton to integral membrane proteins. The main interaction tethering the skeleton to the lipid bilayer is accomplished by ankyrin, which links β-spectrin to band 3 (see Fig. 46–1). The ankyrin binding site is a flexible pocket formed by repeats 14 and 15 of β-spectrin near the C-terminal end of the molecule.38,39 Spectrin also interacts with phosphatidylserine on the inner leaflet of the lipid bilayer.
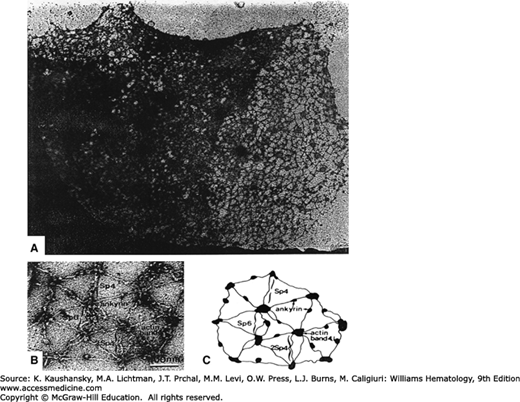
Figure 46–4.
Electron micrograph of the human erythrocyte membrane skeleton. Membrane lipids and transmembrane proteins have been removed and the skeletons were extended during preparation and negative staining to reveal the structure. A. Low-magnification image reveals an ordered network of proteins. B and C. High-magnification image and schematic of the hexagonal lattice showing spectrin tetramers (Sp4) and hexamers (Sp6) or double tetramers (2Sp4). Junctional complexes contain actin filaments and protein 4.1R. Globular ankyrin molecules are bound to spectrin tetramers. (Reproduced with permission from Liu SC et al: Visualisation of the hexagonal lattice in the erythrocyte membrane skeleton. J Cell Biol 1987 Mar;104(3):527-536.)
Nonrepeat sequences in spectrin provide the recognition sites for binding to modifiers, including kinases and calmodulin. The functions of spectrin are to maintain the biconcave disk shape of the red cell, regulate the lateral mobility of integral membrane proteins, and provide structural support for the lipid bilayer.
Erythrocyte ankyrin is encoded by the ANK1 gene, which contains three separate tissue-specific promoters and first exons that are spliced to a common exon 2.40 The 206-kDa protein is a versatile binding partner and has three functional domains: an N-terminal 89-kDa membrane-binding domain, that contains sites for band 3 and other ligands; a central 62-kDa spectrin-binding domain, and a C-terminal 55-kDa regulatory domain that is responsible for the different isoforms of the protein, which influence ankyrin–protein interactions (Fig. 46–5).29
Figure 46–5.
Schematic of human erythrocyte ankyrin. The N-terminal domain consists of 24 ANK repeats, which bind to band 3 and the Rh–RhAg complex. The central domain attaches to spectrin. The C-terminal domain varies in different isoforms of ankyrin, which are produced by alternative splicing of the gene. This domain also contains a conserved death domain of unknown function.
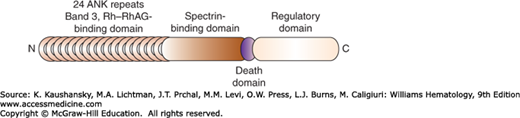
The membrane-binding domain contains 24 tandem ankyrin (ANK) repeats, which are stacked into a superhelical array that is coiled into a solenoid. This structure behaves like a reversible spring, which may contribute to the elasticity of the membrane.22 Each 33-amino-acid ANK repeat is highly conserved and forms an L-shaped structure composed of two antiparallel α helices separated by a β hairpin.41 The ANK repeats are connected by unstructured loops and provide an interface for numerous protein–protein interactions. Erythrocyte ANK repeats specifically bind to band 3 and the Rh–RhAG macromolecular complex.19,29
The spectrin-binding domain contains a small unique subdomain termed ZU5-ANK, which has a β-strand core with several surface loops and binds to β-spectrin through hydrophobic and electrostatic interactions.42 The regulatory domain contains a highly conserved death domain of unknown function in the red cell. The C-terminal section of the regulatory domain varies in the different isoforms of ankyrin, proteins 2.1 to 2.6, which are created by alternative splicing43 and which exhibit different binding affinities for band 3 and spectrin. Phosphorylation of ankyrin reduces binding to band 3 and spectrin tetramers.
The gene encoding protein 4.1 produces diverse isoforms in different tissues and different developmental stages. This diversity is accomplished by the use of alternate first exons under the control of different promoters, and alternate initiation codons. This transcriptional regulation is coupled to complex pre-mRNA splicing events.44,45 The erythrocyte isoform, 4.1R, is produced from the downstream initiation codon and contains exon 16, which encodes an essential part of the spectrin-actin binding domain.
Protein 4.1R is a globular phosphoprotein that contains four structural and functional domains of 30 kDa, 16 kDa, 10 kDa, and 22 to 24 kDa (Fig. 46–6). The N-terminal 30-kDa domain is responsible for binding to the cytoplasmic domains of band 3 and GPC, as well as to p55, thereby linking the skeleton to the lipid bilayer.19 The 10-kDa domain enhances the interaction between spectrin and actin in the junctional complex, which connects spectrin tetramers to each other. The functions of the other two domains are not characterized. Phosphorylation of 4.1R inhibits spectrin–actin–4.1R complex formation and also decreases binding to band 3. Protein 4.1R binds weakly to phosphatidylserine in the lipid bilayer.
Figure 46–6.
Schematic of human erythrocyte protein 4.1R. The protein consists of four domains, with the 30-kDa and the 10-kDa domains involved in binding to other red cell membrane proteins. The C-terminal domain has an asparagine residue at position 502 in isoform 4.1a, which is deamidated in older red cells to form aspartic acid and isoform 4.1b.
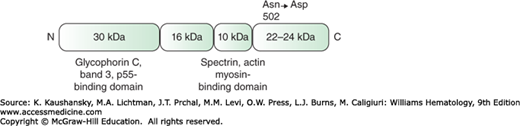
Two forms of 4.1R, a and b, are present in red cells, with protein 4.1b predominating in young erythrocytes. The difference between the two isoforms relates to the gradual deamidation of asparagine 502 to aspartic acid in a nonenzymatic, age-dependent manner, which influences the mobility of the protein on SDS gels.46
Protein 4.2 is a member of the transglutaminase family of proteins,47 but it has no enzyme activity as it lacks the critical triad of residues that form the active transglutaminase site. The exact role of protein 4.2 has not been elucidated, but it stabilizes the link between the skeleton and the lipid bilayer. Protein 4.2 interacts with several proteins, including the cytoplasmic domain of band 3, and this binding site has been identified as a hairpin region toward the center of the protein 4.2 molecule.11,47 Interactions with the ANK repeats in the membrane-binding domain of ankyrin47 and CD47, a component of the Rh complex, have been documented.19,47 In vitro binding studies have revealed an association of protein 4.2 with 4.1R and spectrin. Protein 4.2 binds calcium adjacent to the spectrin-binding loop suggesting that calcium may regulate this interaction. The protein undergoes posttranslational palmitoylation and myristoylation, which suggests an interaction with the lipid bilayer.47
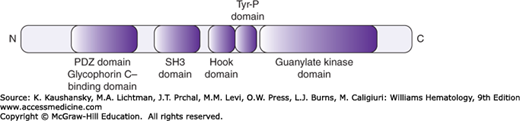
This molecule is a phosphoprotein member of the membrane-associated guanylate kinase (MAGUK) family of proteins.48 In the red cell it is found as part of a ternary complex with GPC and 4.1R and it strengthens the link between the skeleton and the bilayer.19 p55 contains five domains, including an N-terminal PDZ domain, which binds to GPC; an SH3 domain; a central HOOK domain interacting with the 30-kDa domain of 4.1R; a region with tyrosine phosphorylation sites; and a C-terminal guanylate kinase domain (Fig. 46–7).48 The protein is extensively palmitoylated, reflecting an interaction with the membrane bilayer.
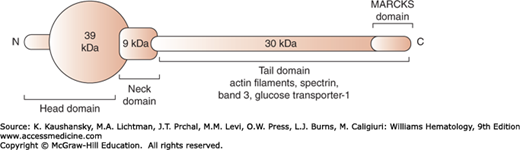
Adducin, a calcium/calmodulin-binding phosphoprotein located at the spectrin–actin junctional complex, is composed of αβ adducin heterodimers, which are structurally similar proteins encoded by separate genes. Adducins contain a 39-kDa globular head region, a small neck region of 9 kDa implicated in oligomerization to form α2β2 heterotetramers, and a 30-kDa cytoplasmic tail with a myristoylated alanine-rich C kinase substrate (MARCKS) phosphorylation domain at the C terminus (Fig. 46–8). The adducin tails cap actin filaments and promote interaction of spectrin and actin.49 They also bind band 3 and the GLUT-1, and thus form part of the macromolecular junctional complex linking the spectrin skeleton to the lipid bilayer (see Fig. 46–1).21,50 The function of adducin is regulated by calcium-dependent calmodulin binding and differential phosphorylation. A primary deficiency of adducin in human disease has not been described; however, mice with targeted inactivation of α– or β-adducin suffer from compensated spherocytic anemia, suggesting that the adducin mutations may be candidates for recessively inherited hemolytic anemia.51
The erythrocyte contains β-type actin assembled into short F-actin protofilaments of 14 to 16 monomers. The length of the filaments is regulated by a “molecular ruler” of two rod-shaped tropomyosin molecules, which are bound along the filament, as well as by two tropomodulin molecules, which cap the filaments at the pointed ends.52 At the barbed end actin is capped by an adducin heterodimer. Dematin or protein 4.9 is a trimeric phosphoprotein, which bundles the actin filaments,53 but also acts as a linker molecule by binding to the transmembrane GLUT-1.21,50
MEMBRANE ORGANIZATION
The structure of the erythrocyte membrane is determined by multiple protein–protein interactions between (1) integral membrane proteins within the lipid bilayer, (2) peripheral proteins in the skeleton, and (3) linker proteins, which tether the skeleton to the transmembrane proteins (see Fig. 46–1). Protein–lipid interactions within the bilayer or between the anionic phospholipids and the underlying membrane skeleton also play a role in cohesion of the membrane components. By using the cytoplasmic domains of embedded proteins as attachment points, the membrane skeleton not only affixes itself to the lipid bilayer but also influences the topology of the transmembrane proteins and constrains their lateral and rotational mobility.
The membrane skeleton resembles a lattice-like network, with approximately 60 percent of the lipid bilayer directly laminated to the underlying skeleton.36 Electron microscopy of stretched membrane skeletons indicate that the individual proteins can be visualized as a highly ordered meshwork of hexagons (see Fig. 46–4).36 The corners of each hexagon consist of a globular macromolecular junctional complex of proteins, including 4.1R and actin, which interact with spectrin tetramers, as well as tropomyosin, tropomodulin, adducin, dematin, and p55.19,21,50 Spectrin tetramers form the arms of the hexagons, cross-bridging individual junctional complexes. These horizontal protein interactions are important in the maintenance of the structural integrity of the cell, accounting for the high tensile strength of the erythrocyte (see Fig. 46–1).
The spectrin/actin skeleton is anchored to the phospholipid bilayer by two major membrane protein complexes: (1) an ankyrin complex that contains transmembrane proteins, band 3, GPA, Rh, and RhAG complex proteins, as well as peripheral proteins ankyrin, protein 4.2, and several glycolytic enzymes, and (2) a distal junctional complex that contains the membrane-spanning proteins band 3, GPC, GLUT-1, Rh, Kell, and XK proteins, in addition to peripheral proteins 4.1R, actin, tropomyosin, tropomodulin, adducin, dematin, and p55. These vertical protein–protein interactions are critical in the stabilization of the lipid bilayer, preventing loss of microvesicles from the cells (see Fig. 46–1).
The avidity of these horizontal and vertical interactions is modulated by posttranslational modifications of the participating proteins, especially phosphorylation. The erythrocyte contains multiple protein kinases and phosphatases that constantly phosphorylate and dephosphorylate specific serine, threonine, and tyrosine residues on band 3, β-spectrin, ankyrin, 4.1R, adducin, and dematin, in a dynamic manner, thereby tightly regulating the structural properties of the membrane. Additionally, membrane protein associations are also influenced by a variety of intracellular factors, including calcium, calmodulin, phosphoinositides, and polyanions such as 2,3-bisphosphoglycerate (BPG). Red cell membrane proteins are also subject to a variety of other posttranslational modifications, including myristoylation, palmitoylation, glycosylation, methylation, deamidation, oxidation, and limited proteolytic cleavage, but the functional effects of these alterations are generally not known.
CELLULAR DEFORMABILITY AND MEMBRANE STABILITY
In performing its primary function of oxygen delivery to the tissues, the erythrocyte has to repeatedly negotiate tiny capillaries in the microvasculature, as well as narrow slits in the spleen, which are much smaller than the diameter of the cell. Consequently, it has to undergo extensive distortion and deformation without fragmentation or loss of integrity, and this property of deformability is critical for survival during its average 120-day life span. The structure of the red cell membrane endows the cell with unique material properties, which makes it highly flexible, yet incredibly resilient, and enables a very rapid response to circulatory shear stress.
Elegant biophysical studies have identified three features that regulate the deformability of the cell: (1) the biconcave disk shape, which reflects the cell surface-area-to-volume ratio; (2) the viscoelastic properties of the membrane, which depend on the structural and functional integrity of the membrane skeleton; and (3) the cytoplasmic viscosity, which is determined primarily by intracellular haemoglobin.54
The unique biconcave disk shape of the erythrocyte provides a high ratio of surface area to cellular volume and this excess of membrane is critical for survival of the cell. It enables the red cell to stretch and distort when it passes through the microcirculation and protects it from destruction. To maintain the shape of the cell and to prevent loss of membrane microvesicles, the lipid bilayer and the skeleton have to be in direct contact with each other. The cohesion between the two sections of the membrane depends on protein–protein interactions between transmembrane proteins and peripheral proteins in the vertical plane of the membrane. These contacts are represented by the two macromolecular complexes (ankyrin–band 3 complex and the junctional complex) anchoring the skeleton to the integral proteins. To prevent fragmentation of the membrane and loss of the biconcave disk shape, the structural integrity of the membrane skeleton is critical. In this regard, the horizontal interactions of the peripheral proteins of the junctional complex, mainly 4.1R and actin, which link the tail ends of the spectrin tetramers together, is a major determinant of membrane stability. Spectrin heterodimer self-association, which links the head regions of the spectrin tetramers, is also of paramount importance.
The viscoelastic properties of the membrane are intrinsic features of the spectrin skeleton. The enormous distortion imposed on the cell during passage through the microvasculature is accommodated by the dynamic dissociation of spectrin tetramers into dimers, and subsequent reassociation to restore the original shape once the shear stress is removed.55 The lattice structure of the skeleton facilitates this flexibility, as the individual hexagons are either in a compact configuration, with the junctional complexes close to each other and the spectrin tetramers coiled between them, or in an extended configuration, which allows large unidirectional deformation without disruption of the skeleton (see Fig. 46–4). The structure of the spectrin repeats also play a major role in the elasticity of the skeleton. Each triple helical repeat behaves partly as an independently folding unit and has a different thermal stability.56 Cysteine labeling studies indicated that shear stress forced the unfolding of the least stable repeats.57 These studies highlight the flexibility of the spectrin repeats and support the concept that their unfolding and refolding contributes to the deformability of the membrane. In addition, the elasticity of the ANK repeats may also facilitate the dynamic changes in the membrane during circulatory shear stress.22
Red cell viscosity is largely determined by the concentration of intracellular hemoglobin, which is tightly regulated to minimize cytoplasmic viscous dissipation during cellular deformation. As the mean cell hemoglobin concentration rises above 37 g/dL, the viscosity increases exponentially, and this compromises the deformability of the cell under increased circulatory shear stress.54 The hemoglobin concentration is critically dependent on red cell volume, which is primarily determined by the total cation content of the cell. Numerous membrane pumps and ion channels regulate the transport of sodium and potassium across the membrane (Fig. 46–9).
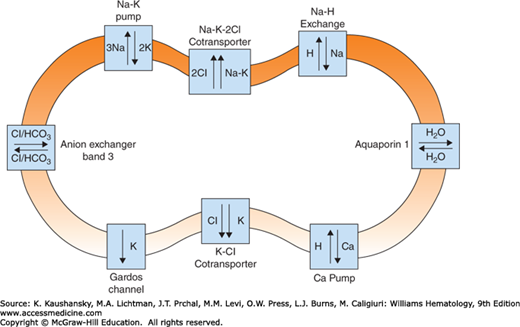
MEMBRANE PERMEABILITY
The red cell membrane displays selective permeability to cations and anions and it maintains a high potassium, low sodium, and very low calcium content within the cell.58 Ion transport pathways in the red cell membrane (see Fig. 46–9) include energy-driven membrane pumps, gradient-driven systems, and various channels. Several transport mechanisms exist for cations, including two energy-driven pumps.58 The sodium pump is a Na+K+ adenosine triphosphatase (ATPase) that extrudes three sodium ions in exchange for two potassium ions entering the red cell. Calcium is pumped out of the cell by a calmodulin-activated Ca2+ ATPase, which protects the cell from deleterious effects of calcium, such as echinocytosis (Chap. 31), membrane vesiculation, calpain activation, membrane proteolysis, and cellular dehydration.58 The Ca2+-activated K+ channel, also called the Gardos channel, causes selective loss of K+ in response to increased intracellular Ca2+. The Na+K+ gradient established by the sodium pump is used by several passive, gradient-driven systems to move ions across the red cell membrane.58 The systems include the K+Cl– cotransporter, the Na+K+2Cl– cotransporter, and the Na+H+ exchanger.
Chloride and bicarbonate anions are readily exchanged through band 3. The red cell is highly permeable to water, which is transported by aquaporin-1 (AQP1),59 and glucose is taken up by the glucose transporter.60 The membrane also contains an ATP-driven oxidized glutathione (GSSG) transporter and amino acid transport systems.58 Larger charged molecules, such as ATP, do not cross the membrane.
RED CELL MEMBRANE DISORDERS
Hemolytic anemias resulting from defects in the erythrocyte membrane comprise an important group of hereditary anemias. The disorders are characterized by altered red cell morphology, which is reflected in the nomenclature of HS, HE, hereditary pyropoikilocytosis (HPP) and southeast Asian ovalocytosis (SAO), which are the most common disorders in this group. Protein studies have identified the underlying membrane abnormalities and advances in molecular biology have enabled further characterization of these disorders and, in many cases, identification of the causative mutations. These molecular analyses have provided additional information on the pathogenesis of these disorders and important insights into the structure–function relationships of erythrocyte membrane proteins.
As predicted in 1984 by Jiri Palek61 and confirmed by subsequent studies, protein defects that compromise vertical interactions between the membrane skeleton and the lipid bilayer result in destabilization of the bilayer, loss of membrane microvesicles and spherocyte formation; whereas mutations affecting horizontal protein interactions within the membrane skeletal network disrupt the skeleton resulting in defective shape recovery and elliptocytes (Table 46–2). Red cell membrane disorders exhibit significant heterogeneity in their clinical, morphologic, laboratory and molecular characteristics.
| Protein | Disorder | Comment |
|---|---|---|
| Ankyrin | HS | Most common cause of typical dominant HS |
| Band 3 | HS, SAO, NIHF, HAc | “Pincered” HS spherocytes seen on blood film presplenectomy; SAO results from 9-amino-acid deletion |
| β-Spectrin | HS, HE, HPP, NIHF | “Acanthocytic” spherocytes seen on blood film presplenectomy; location of mutation in β-spectrin determines clinical phenotype |
| α-Spectrin | HS, HE, HPP, NIHF | Location of mutation in α-spectrin determines clinical phenotype; α-spectrin mutations most common cause of typical HE |
| Protein 4.2 | HS | Primarily found in Japanese patients |
| Protein 4.1 | HE | Found in certain European and Arab populations |
| GPC | HE | Concomitant protein 4.1 deficiency is basis of HE in GPC defects |
Hereditary spherocytosis is characterized by the presence of osmotically fragile spherical red blood cells on the blood film (Fig. 46–10B). The disorder was first described in 1871 as microcythemia in a case history by two Belgian physicians.62
Figure 46–10.
Blood films from patients with erythrocyte membrane disorders. A. Normal blood film. B. HS with dense spherocytes. C. SAO with large ovalocytes exhibiting a transverse ridge. D. HE with elongated elliptocytes and some poikilocytes. E. HSt with cup-shaped stomatocytes. F. Hereditary abetalipoproteinemia with acanthocytes. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
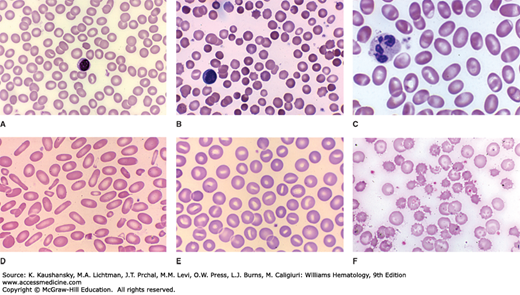
HS occurs in all racial and ethnic groups. It is the most common inherited hemolytic anemia in individuals of northern European ancestry, affecting approximately 1 in 2000 individuals in North America and Europe.63 It is also common in Japan and in Africans from southern Africa. Males and females are affected equally.
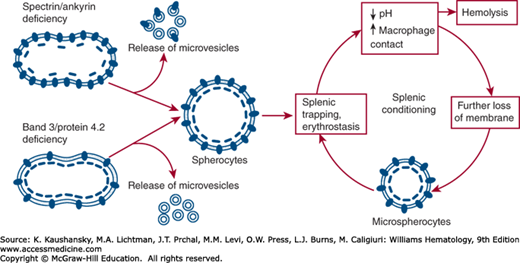
The hallmark of HS erythrocytes is loss of membrane surface area relative to intracellular volume, which accounts for the spherical shape and loss of central pallor of the cell (Figs 46-10B and 46-11C). Spherocytes exhibit decreased deformability and are thus selectively retained, damaged and ultimately destroyed in the spleen, which causes the hemolysis experienced by HS patients. The HS red cell membrane is destabilized by a deficiency of critical membrane proteins, including spectrin, ankyrin, band 3 and protein 4.2, which decreases the vertical interactions between the skeleton and the bilayer, resulting in the release of microvesicles and loss of surface area (Fig. 46–12). It is hypothesized that two mechanisms underlie the membrane loss: (1) in cells with spectrin/ankyrin deficiency, sections of the lipid bilayer and band 3 are not in contact with the skeleton, which will increase the lateral and rotational mobility of band 3, allowing lipid microvesicles containing band 3 to be generated, and (2) in cells with decreased amounts of band 3/protein 4.2, the stabilizing effect of the transmembrane section of band 3 on the lipid bilayer is lost, facilitating the formation of band 3-free microvesicles.13
Figure 46–11.
Scanning electron micrographs of erythrocytes with abnormal morphology due to membrane defects. A. Normal discocyte. B. Echinocyte. C. Spherocyte. D. Stomatocytes. E. Ovalocytes. F. Elliptocytes. G. Acanthocytes. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
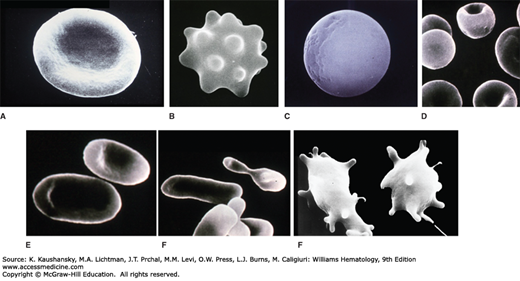
Figure 46–12.
Pathobiology of hereditary spherocytosis (HS). The primary defect in HS is a deficiency of one of the membrane proteins, which destabilizes the lipid bilayer and leads to a loss of membrane in the form of microvesicles. This reduces the surface area of the cell and leads to spherocyte formation. Red cells with a deficiency of spectrin or ankyrin produce microvesicles containing band 3, whereas a reduced amount of band 3 or protein 4.1R gives rise to band 3–free microvesicles. Spherocytes have decreased deformability and are trapped in the spleen where the membrane is further damaged by splenic conditioning, which ultimately results in hemolysis.
Analysis of HS red cell membrane proteins by several research groups has revealed quantitative abnormalities of spectrin, ankyrin, band 3, and protein 4.2 in 70 to 90 percent of the cases.13,63,64 This spectrum of defects is found worldwide in all the HS cohorts that have been studied; however, the relative frequency of each defect varies with the geographical area and ethnic group. In the United States, parts of Europe, and in Korea, the most common defect is ankyrin deficiency (30 to 60 percent),63,65,66 whereas it is relatively uncommon elsewhere in the world (<15 percent). In other parts of Europe64,67 and in South Africa (unpublished), band 3 deficiency is the main defect. In Japan, almost half of the HS cases are caused by a decreased amount of protein 4.2, and in Korea and South Africa, this defect is the second most common, but in other populations it is rare (<6 percent).63,64,65 The underlying gene mutations have not been investigated in all HS subjects, but the limited research that has been conducted on the defective genes has identified more than 140 different mutations, which are often unique to a family.
Concomitant ankyrin and spectrin deficiency was first described in two patients with severe atypical HS and the primary defect was identified as an ankyrin abnormality.68 Subsequent DNA analysis of the ANK1 gene in patients with typical HS identified several mutations,69 and numerous other studies have shown that ankyrin/spectrin deficiency is a common cause of HS. Ankyrin binds to spectrin with high affinity and attaches it to the membrane, which stabilizes the molecule. Because ankyrin is present in limiting amounts, a deficiency of ankyrin causes an equivalent loss of spectrin.
Different types of ankyrin mutations have been identified throughout the gene, indicating that there are several mechanisms that ultimately result in a decreased amount of ankyrin in the membrane. Interestingly, the majority of these mutations are frameshift and nonsense mutations that either result in unstable transcripts that are destroyed by nonsense-mediated mRNA decay or else produce a truncated defective ankyrin molecule.66 More than 50 mutations have been documented and they are typically family-specific, although a few recurrent mutations have been described69,70 and 15 to 20 percent of mutations are de novo.63 Missense mutations have been documented in all the ankyrin domains and are thought to disrupt normal ankyrin–protein interactions. A few splicing mutations have been identified, including a mutation in intron 16, which created a new splice acceptor site and a complex pattern of aberrant splicing.71 Both parents were heterozygous for this mutation and the proband was homozygous, indicating that homozygosity for an ankyrin mutation is compatible with life.
Mutations in the erythroid-specific promoter of the ANK1 gene are common in recessive HS. A dinucleotide deletion impairs the binding of a transcription factor complex, which leads to a reduced number of ankyrin transcripts.72 Point mutations in a barrier insulator element of the promoter also decrease transcription of the gene.73
Cytogenetic studies have identified a few ankyrin-deficient HS patients with a contiguous gene syndrome that includes deletion of the ankyrin gene locus at 8p11.2. These patients additionally suffer from dysmorphic features, psychomotor retardation, and hypogonadism.74
A subset of HS patients present with a band 3 deficiency, typically accompanied by a secondary decrease in protein 4.2, a result of the reduction in protein 4.2 binding sites in the cytoplasmic domain of band 3. The extent of band 3 deficiency in heterozygous patients ranges between 20 and 50 percent, depending on the severity of the mutation, and the compensatory effect of the in trans normal allele. Mushroom-shaped “pincered” cells are commonly seen on the blood film of HS patients with a band 3 abnormality.
More than 55 underlying mutations have been described; they are variable and occur throughout the band 3 gene.13,66 Null mutations are typically family-specific and are caused by frameshift or nonsense mutations, or, in a few cases, by abnormal splicing, all of which result in truncated nonfunctional proteins or unstable transcripts that are not translated into protein. Missense mutations are common and often occur in several kindred. Highly conserved arginine residues at the internal boundaries of the transmembrane segments of the protein (see Fig. 46–2) are frequently mutated, including residues 490, 518, 760, 808, and 870.66,75 The mutations probably interfere with the cotranslational insertion of band 3 into the endoplasmic reticulum and ultimately into the red cell membrane. Short in-frame insertions or deletions have been documented and presumably also impair insertion of the mutant protein into the lipid bilayer.
Mutations in the cytoplasmic domain of band 3 impact on the interaction of band 3 with proteins in the membrane skeleton, or may alter the conformation of the protein rendering it unstable and prone to degradation prior to insertion into the membrane. Some cytoplasmic mutations, such as band 3 Cape Town and band 3 Mondega, are silent in the heterozygous state, but exacerbate the clinical presentation when inherited in trans to another mutation.76,77
Erythrocytes from HS patients with defects in spectrin or ankyrin are deficient in spectrin. The degree of deficiency correlates with the severity of hemolysis, the response to splenectomy and the ability to withstand mechanical shear stress.78,79 Visualization of the membrane skeleton of these red cells revealed a decreased density of the spectrin filaments connecting the junctional complexes.80 The causative mutations occur in either α– or β-spectrin genes.
Defects in α-spectrin are rare and are associated with severe recessive HS. During erythropoiesis α-spectrin is synthesized in a two- to fourfold excess over β-spectrin and heterozygotes thus still produce sufficient α-spectrin to form heterodimers with all the β-spectrin molecules, which will not result in spectrin deficiency. The defect will only be manifested in individuals who are homozygous or doubly heterozygous for mutations in α
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


