Thrombocytosis is a common clinical problem frequently encountered during routine evaluation. The diagnostic workup entails a step-by-step approach, which allows for an accurate assessment of the underlying cause. A thorough clinical history and physical examination may help differentiate thrombocytosis secondary to a reactive process versus an underlying clonal proliferation process. Once essential thrombocytosis is evident, relevant laboratory evaluation for an ongoing myeloproliferative disorder is paramount. Various treatment modalities have been proven to be beneficial. With further scientific investigation underway, molecular therapies may soon be cornerstones of therapy in essential thrombocytosis.
Thrombocytosis is defined as having excess platelets in the blood. The normal platelet count in adults ranges between 150,000 and 450,000/μL (mean ± 2 standard deviations), regardless of sex and ethnicity. Based on the normal distribution, this implies that a platelet count exceeding 450,000/μL exists in about 2.5% of the population.
An increased platelet count usually has an acquired cause. It is rarely congenital. Congenital thrombocytosis results from gain-of-function mutations in either thrombopoietin (TPO) or its receptor (MpL). The altered regulation is usually transmitted in an autosomal dominant fashion and often involves the 5′-untranslated region of the TPO mRNA (a donor splice site), resulting in increased translational efficiency. Increases in serum TPO concentrations are noted in those patients with a TPO gene mutation.
Acquired thrombocytosis may be either a primary or secondary process. Essential thrombocytosis (ET) is also known as primary thrombocytosis, essential thrombocythemia, and autonomous thrombocytosis. It is a disease of the bone marrow associated with myeloproliferative neoplasms that lead to an increase in platelets. This process may be growth factor independent or growth factor hypersensitive. Acquired thrombocytosis may be reactive and secondary to an unrelated condition such as infection, chronic inflammation, hemolysis, iron deficiency anemia, or splenectomy, and is referred to as reactive thrombocytosis (RT).
Reactive thrombocytosis (RT)
Prevalence and Relevance
RT refers to an increase in platelet count associated with conditions other than chronic myeloproliferative or myelodysplastic disorders. It is observed in a variety of conditions that may cause an acute, transient, or sustained increase in platelet counts ( Table 1 ). It is generally accompanied by the signs and symptoms of the underlying disease and normalizes, or is expected to normalize, after resolution of this condition. In routine clinical practice, RT accounts for more than 85% of cases of thrombocytosis, even in patients with extreme thrombocytosis (platelet count >1000 × 10 3 /μL).
| Condition | Adults (n = 777) | Platelet Count of 1 Million/μL or More (n = 280) | Children (n = 663) |
|---|---|---|---|
| Infection | 22 | 31 | 31 |
| Rebound thrombocytosis | 19 | 3 | 15 |
| Tissue damage (eg, surgery) | 18 | 14 | 15 |
| Chronic inflammation | 13 | 9 | 4 |
| Malignancy | 6 | 14 | 2 |
| Renal disorders | 5 | NS | 4 |
| Hemolytic anemia | 4 | NS | 19 |
| After splenectomy | 2 | 19 | 1 |
| Blood loss | NS | 6 | NS |
| Primary thrombocythemia | 3 | 14 | 0 |
It is important to distinguish between RT and ET because of their different clinical manifestations and treatment strategies. There is a well-established association of ET with vasomotor symptoms and thrombotic or bleeding complications. In general, thromboembolic complications are rare in RT compared with ET, unless clinically provoked by underlying conditions such as malignancy or atherosclerosis. More often, high-risk patients with ET require cytoreductive therapy to prevent catastrophic thrombohemorrhagic complications, whereas the prothrombotic potential of RT may be too low to even justify the use of platelet directed therapy, even in the context of surgical procedures. Because of the low likelihood of vascular complications, there is no evidence to support the use of cytoreductive agents in patients with RT.
Pathogenesis
A list of the causes of RT is shown in Table 1 . Several cytokines and lymphokines are increased in the blood of patients with RT, especially those associated with infection, inflammation, malignancy, and tissue damage. These cytokines include interleukin (IL)-6, thrombopoietin, IL-1, IL-4, interferon γ, and tumor necrosis factor-α. Of all the cytokines implicated, the most compelling evidence for their pathogenic role is noted with IL-6 and interferon γ. The administration of a transcription factor that mimics interferon γ led to the correction of thrombocytopenia in a genetic model. IL-6 stimulates TPO production in the liver. Thrombocytosis and induction of other acute phase reactants are noted with the administration of human recombinant IL-6 in patients with metastatic cancer.
RT is a predictable finding after splenectomy, with an incidence approaching 50% in large series. Thrombocytosis is commonly seen immediately after splenectomy and normalizes in most cases within several months, and rarely only after many years. Hyposplenism-associated RT may reflect platelet redistribution in the peripheral blood as well as altered metabolism of thrombopoietic cytokines. Persistent thrombocytosis has been described in congenital and functional asplenia (celiac sprue, amyloidosis). Whatever the cause, in the absence of a chronic myeloproliferative disorder (MPD), hyposplenic-associated thrombocytosis is rarely associated with an increased risk of thrombosis.
Reactive Thrombocytosis Versus Essential Thrombocytosis (ET)
ET is a clonal proliferation process with subsequent increase in platelet counts. A thorough history and physical examination are the most important elements in differentiating RT from ET. Determining the duration of thrombocytosis is a key diagnostic step of the initial workup. If no obvious explanation is provided, longstanding and persistent thrombocytosis strongly suggests ET. A history of vasomotor symptoms, thrombohemorrhagic complications, and physical examination findings such as splenomegaly and acral erythema also strongly suggest ET. An increased platelet count in the presence of conditions associated with RT, as outlined in Table 1 , may favor the diagnosis of RT. However, if thrombocytosis is noted in the absence of associated clinical conditions such as vasculitis and infection, then ET may be more likely.
Complete blood count with differential counts, red blood cell indices, and peripheral blood smear examination, as well as iron studies, provide additional information for differentiating ET from RT ( Fig. 1 ). Laboratory findings that suggest RT include microcytosis, which may indicate iron deficiency anemia. Although a normal ferritin level may reduce the likelihood of iron deficiency anemia associated with RT, a low level does not necessarily eliminate the possibility of ET. Increased acute phase reactants such as C-reactive protein, erythrocyte sedimentation rate, plasma fibrinogen, and IL-6 levels have been shown to be increased in RT. The presence of Howell-Jolly bodies on the peripheral blood smear suggests hyposplenism, supporting a diagnosis of RT, whereas a leukoerythroblastic smear suggests primary myelofibrosis (PMF) (also known as myelofibrosis with myeloid metaplasia).
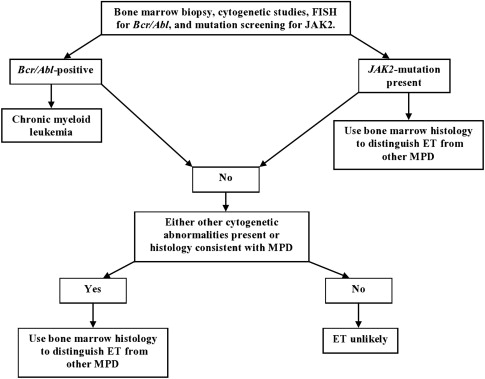
It is difficult to discriminate between ET and RT based on the degree of thrombocytosis, regardless of how high the platelet counts might be. There is no diagnostic usefulness of platelet indices (mean volume, size distribution width) in discriminating between ET and RT because of the considerable overlap in the measured values. Although abnormalities of platelet function as measured by prolonged bleeding time, decreased adenosine triphosphate secretion, and altered thromboxane generation are seen in ET, these laboratory assays are not routinely used because they require substantial expertise to perform and interpret.
The presence of cytogenetic abnormalities, such as the Janus kinase 2 (JAK2 V617F ) mutation, is strongly associated with MPDs. This abnormality has not been detected in healthy controls or in patients with secondary erythrocytosis or RT, thus making mutation screening an important part of the diagnostic workup (see Fig. 1 ). Although clonal cytogenetic abnormalities can be detected and are diagnostic of ET, not all causes of ET have detectable cytogenetic abnormalities (<5% of patients with ET).
Consequently, bone marrow histology evaluation is still a critical part of the evaluation process. Although the bone marrow histology is normal in RT, it displays a wide variety of abnormalities in ET depending on the cause of the underlying chronic myeloproliferative disease (CMD). These abnormalities include aberrations in cellular morphology, increased numbers of megakaryocytes, presence of megakaryocyte clusters, and reticulin fibrosis.
Essential thrombocytosis (ET) and myeloproliferative states
ET is a clonal expansion of a multipotential stem cell observed in the presence of a chronic MPD or myelodysplastic syndrome (MDS). Clonal or autonomous thrombocytosis is part of ET and is seen in 50% of patients with polycythemia vera (PV). It is also seen in about 35% of patients with chronic myeloid leukemia (CML).
These disorders (MPD and MDS) may be classified as those that exhibit trilineage morphologic dysplasia (in MDS) and those that do not (in MPD). The classification of chronic myeloid disorders is shown in Box 1 . Although thrombocytosis in MDS has been associated with the presence of ringed sideroblasts and may be linked with certain cytogenetic abnormalities including trisomy 8, 5q− syndrome, and abnormalities of chromosome 3, the underlying pathogenic connection between these defects and thrombocytosis is unclear. Except for a few atypical myelodysplasia states and CML, the molecular pathogenesis of the MPDs is also unclear.
- 1.
MPS
- 2.
MPDs
- a.
Classic MPDs
- i.
Molecularly defined
- 1.
CML ( Bcr / Abl + )
- 1.
- ii.
Clinicopathologically assigned (Bcr/Abl − and frequently associated with JAK2 V617F mutation)
- 1.
Essential thrombocythemia
- 2.
PV
- 3.
Myelofibrosis with myeloid metaplasia
- 1.
- i.
- b.
Atypical MPDs
- i.
Molecularly defined
- 1.
PDGFRA -rearranged eosinophilic/mast cell disorders (eg, FIP1L1-PDGFRA )
- 2.
PDGFRB -rearranged eosinophilic disorders (eg, TEL / ETV6 – PDGFRB )
- 3.
Systemic mastocytosis associated with c-kit mutation (eg, c-kit D 816 V )
- 4.
8p11 Myeloproliferative syndrome (eg, ZNF198 / FIM / RAMP-FGFR1 )
- 1.
- ii.
Clinicopathologically assigned
- 1.
Chronic neutrophilic leukemia
- 2.
Chronic eosinophilic leukemia, molecularly not defined
- 3.
Hypereosinophilic syndrome
- 4.
Chronic basophilic leukemia
- 5.
Chronic myelomonocytic leukemia
- 6.
Juvenile myelomonocytic leukemia (associated with recurrent mutations of RAS signaling pathway molecules including PTPN11 and NF1 )
- 7.
Systemic mastocytosis, molecularly not defined
- 8.
Unclassified MPD
- 1.
- i.
- a.
Essential thrombocytosis (ET) and myeloproliferative states
ET is a clonal expansion of a multipotential stem cell observed in the presence of a chronic MPD or myelodysplastic syndrome (MDS). Clonal or autonomous thrombocytosis is part of ET and is seen in 50% of patients with polycythemia vera (PV). It is also seen in about 35% of patients with chronic myeloid leukemia (CML).
These disorders (MPD and MDS) may be classified as those that exhibit trilineage morphologic dysplasia (in MDS) and those that do not (in MPD). The classification of chronic myeloid disorders is shown in Box 1 . Although thrombocytosis in MDS has been associated with the presence of ringed sideroblasts and may be linked with certain cytogenetic abnormalities including trisomy 8, 5q− syndrome, and abnormalities of chromosome 3, the underlying pathogenic connection between these defects and thrombocytosis is unclear. Except for a few atypical myelodysplasia states and CML, the molecular pathogenesis of the MPDs is also unclear.
- 1.
MPS
- 2.
MPDs
- a.
Classic MPDs
- i.
Molecularly defined
- 1.
CML ( Bcr / Abl + )
- 1.
- ii.
Clinicopathologically assigned (Bcr/Abl − and frequently associated with JAK2 V617F mutation)
- 1.
Essential thrombocythemia
- 2.
PV
- 3.
Myelofibrosis with myeloid metaplasia
- 1.
- i.
- b.
Atypical MPDs
- i.
Molecularly defined
- 1.
PDGFRA -rearranged eosinophilic/mast cell disorders (eg, FIP1L1-PDGFRA )
- 2.
PDGFRB -rearranged eosinophilic disorders (eg, TEL / ETV6 – PDGFRB )
- 3.
Systemic mastocytosis associated with c-kit mutation (eg, c-kit D 816 V )
- 4.
8p11 Myeloproliferative syndrome (eg, ZNF198 / FIM / RAMP-FGFR1 )
- 1.
- ii.
Clinicopathologically assigned
- 1.
Chronic neutrophilic leukemia
- 2.
Chronic eosinophilic leukemia, molecularly not defined
- 3.
Hypereosinophilic syndrome
- 4.
Chronic basophilic leukemia
- 5.
Chronic myelomonocytic leukemia
- 6.
Juvenile myelomonocytic leukemia (associated with recurrent mutations of RAS signaling pathway molecules including PTPN11 and NF1 )
- 7.
Systemic mastocytosis, molecularly not defined
- 8.
Unclassified MPD
- 1.
- i.
- a.
ET
Epidemiology
ET is a diagnosis of exclusion once other causes of thrombocytosis such as reactive conditions, MDS, and other myeloproliferative states have been eliminated (see Table 1 ). Among the classic MPDs, ET has the highest prevalence (about 24/100,000) and carries the best prognosis. ET does not routinely occur in children. The median age at diagnosis is 40 to 60 years. There is a suggestion of a female preponderance.
Distinction from Other Causes of ET
Because of MDS, PMF, and CML can all mimic ET in their presentation, it is important to clinically distinguish these separate entities when a working diagnosis of ET is considered. Workup should include exclusion of CML by conventional cytogenetics and fluorescence in situ hybridization (FISH) testing for BCR/ABL. Bone marrow histology should be examined for features that suggest MDS and PMF, such as the presence of trilineage dysplasia and increased marrow cellularity with atypical megakaryocyte hyperplasia and myelofibrosis, respectively.
Pathophysiology
Before the discovery of the JAK2 V617F mutation, scant information existed regarding the molecular pathogenesis of ET. This new discovery has advanced knowledge of the pathophysiology of the condition, as well as the development of new therapeutic tools. The JAK2 V617F mutation, which rarely exists in a homozygous state, is estimated to occur at a frequency of 23% to 60% in ET and 95% in PV. JAK2 V617F is part of the cytoplasmic protein tyrosine kinases. This mutation is an acquired G to T nucleotide shift at position 1849 in exon 12, which leads to a valine to phenylalanine substitution at codon 617 (JAK2 V617F ). This mutation is located in the pseudokinase domain (JH2) of the JAK2 protein and interferes with autoinhibitory function. Consequently, mutated JAK2 exists in a constitutively phosphorylated state.
JAK2 V617F mutation has been associated with induction of erythropoietin hypersensitivity, a hallmark of PV and other classic MPDs. In vivo, a retrovirus containing JAK2 V617F , when transduced into murine bone marrow, induced erythrocytosis in the transplanted mice. These findings suggest some pathogenic relevance for this particular mutation in MPDs.
Clinical Features of ET
The diagnosis of ET is usually incidental. Many patients with ET are asymptomatic at diagnosis and have a prolonged, stable, or uneventful clinical course. The signs and symptoms may be non–life threatening, such as the presence of vasomotor features (headaches, erythromelalgia, and lightheadedness), splenomegaly, or hypertension, or life threatening, such as thrombohemorrhagic complications.
Erythromelalgia
Erythromelalgia is a classic vasomotor symptom of essential thrombocythemia. It has been described even in patients with only marginal increase in platelet counts. It is defined as a burning sensation associated with a red discoloration of the hands or feet. This condition can be precipitated by heat or exercise. The ensuing digital ischemic changes may be caused by arteriolar inflammation secondary to abnormal platelet-endothelium interaction. Histopathologic studies in erythromelalgia have shown platelet-rich arteriolar microthrombi with endothelial inflammation and intimal proliferation. The prompt response of erythromelalgia to aspirin (40–325 mg/d) is consistent with a diagnosis of a vasomotor symptom as opposed to a thrombotic complication.
Thrombosis
Age greater than 60 years, thrombotic history, leukocytosis greater than 11×10 3 /μL, and presence of the JAK2 V617F mutation are considered important factors in assessing the risk of thrombosis. In general, thrombotic events are more frequent than bleeding episodes, with more arterial events than venous events. Tables 2 and 3 list the incidence of thrombotic and hemorrhagic events in ET, respectively. It is common to have both microcirculatory and large vessel involvement. Abdominal large vessel occlusions and cerebral sinus thrombosis are potentially catastrophic events that occasionally occur in patients with ET.
| n | Platelet × 10 9 /L(Median/Mean) | Asymptomatic (%) | Major Thrombosis (%) | Major Arterial Thrombosis a (%) | Major Venous Thrombosis a (%) | MVD (%) | Total Bleeds (%) (Major) | |
|---|---|---|---|---|---|---|---|---|
| ET | ||||||||
| Bellucci et al, 1986 | 94 | 1200 | 67 | 22 | 81 | 19 | 43 | 37 (3.2) |
| Fenaux et al, 1990 | 147 | 1150 | 36 | 18 | 83 | 17 | 34 | 18 (4) |
| Cortelazzo et al, 1990 | 100 | 1135 | 34 | 11 | 91 | 9 | 30 | 9 (3) |
| Colombi et al, 1991 | 103 | 1200 | 73 | 23.3 | 87.5 | 12.5 | 33 | 3.6 (1.9) |
| Basses et al, 1999 | 148 | 898 | 57 | 25 | NA | NA | 29 | 6.1 (NA) |
| Jensen et al, 2000 | 96 | 1102 | 52 | 14 | 85 | 15 | 23 | 9 (5.2) |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


