Whole-genome and Whole-exome Sequencing in Hereditary Cancer: Impact on Genetic Testing and Counseling
Julianne M. O’Daniel
Kristy Lee
The past few years have seen a whirlwind of technologic advances in terms of genetic testing. Tumbling laboratory costs have placed whole-genome sequencing (WGS) and whole-exome sequencing (WES) at the forefront of new genetic technologies, gaining attention from clinicians and health care administrators who want to tap into these cutting-edge technologies for their patients. Similar to all new health technologies, WGS and WES present both challenges and opportunities. In this chapter, we hope to present a practical examination of these new technologies in the application to hereditary cancer.
Whole-Genome Sequencing/Whole-Exome Sequencing Technologies
Although there are several different technology platforms, the basic premise of current sequencing technology, often referred to as “next-generation” sequencing is to determine the base sequences of huge numbers of DNA segments all performed in parallel, which are then typically aligned to a genomic reference in order to detect genetic variation.1 In both WGS and WES, the DNA sample is first sheared randomly into small fragments, the length of which may vary based on the sequencing platform. Since the original sample contains multiple copies of genomic DNA, the random shearing results in the same segment of DNA being fragmented in different ways. This is important for the alignment step below. These fragments are then amplified through a polymerase chain reaction step similar to traditional sequencing. The result is a library containing hundreds of copies of each of the fragments.
At this stage, WES requires two additional steps to enable focused analysis of the exome, which represents less than 2% of the human genome. The library is enriched for the exonic regions by using oligonucleotide probes that hybridize to, or capture, the specified exon targets.2 The uncaptured DNA fragments are washed away, and an amplification step follows to maximize the amount of captured exonic fragments.
Several kits are commercially available to capture the exome in this fashion. When choosing a kit or testing laboratory, it is important to consider how the exonic regions are defined and covered by the hybridization probes as that will affect the purity and completeness of your exome coverage.2
Several kits are commercially available to capture the exome in this fashion. When choosing a kit or testing laboratory, it is important to consider how the exonic regions are defined and covered by the hybridization probes as that will affect the purity and completeness of your exome coverage.2
The next step for both WGS and WES is the concurrent sequencing of the whole library or the enriched library, respectively (massively parallel sequencing). Dependent on the technology-specific chemistry, the sequencing instrument uses the library fragments to determine the sequence, which is then captured base-by-base by the instrument.
At this stage, quality scores are also calculated pertaining to both individual and sequences of base calls. These scores are frequently reported as Q scores and represent the logarithmic chance that the call is incorrect. For example, Q20 equates to a 1 in 100 chance the call is wrong, Q30 equates to a 1 in 1,000 chance, and Q40 is approximately a 1 in 10,000 chance. The result from this step is a digital file of short sequences, or reads, with their quality scores called the fastq file.
The reads must then be aligned computationally to a human reference genome to produce an assembly of the individual’s genome or exome sequence (Fig. 2.1). Using a reference sequence to guide assembly is referred to as “resequencing,” as opposed to de novo sequencing, which does not align to a known reference. In most cases, the publicly available human genome reference sequence is used. Since the fragments were randomly sheared, a number of reads should align to most of the bases of the reference. This overlap helps ensure accurate alignment and variant identifications. The number of times a specific reference base position is matched with a base in the
aligned reads is called the depth of coverage. In other words, if five reads overlap the base position, then the coverage is 5× at that position.
aligned reads is called the depth of coverage. In other words, if five reads overlap the base position, then the coverage is 5× at that position.
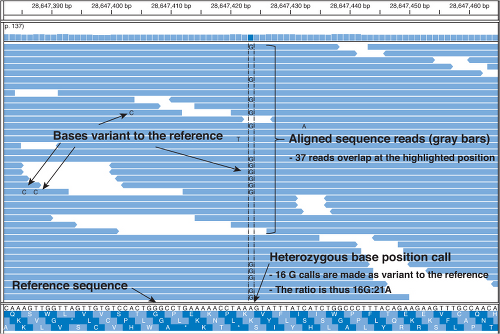 Figure 2.1. Computer display of aligned sequence reads at a given position in the genome. |
A separate algorithm considers the base calls and quality scores of all reads overlapping a specific position in the reference sequence to make a consensus base call at that location in the individual’s genome. For example, if the five overlapping reads all have a G at that position, then the call would be a homozygous G/G. However, if two of the reads have a T and three have a G, then it might mean the individual is heterozygous T/G at that position, or it may be that the T’s (or G’s for that matter) are incorrect. The higher the depth of coverage and the higher the quality of the individual base calls, the higher the confidence that the base called at that position is correct.3 Higher coverage is imperative for determining heterozygous calls or when low levels of mosaicism may be important, such as in tumor samples. The generally reported coverage for clinical WGS/WES testing ranges from a genome-wide average coverage of approximately 30 to 80 times with WES at the higher end of the spectrum. The enrichment/capture step for WES does not perform at a uniform efficiency across the entire exome, leading to a broader spread of coverage depths as compared with WGS, and thus the need for higher average depth of coverage.2
The final and arguably most complicated step is that of clinical interpretation. At this stage, the identified variants are examined against available databases to determine their functional impact and possible clinical significance.4,5 These analyses generally require automated searches initially to collect frequency data and functional consequence predictions as well as comparisons to reported pathogenic variants in clinical databases such as the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk). Following the automated annotation; however, a manual review is essential to identify which variants may be truly pathogenic based on available clinical evidence often collected in multiple locus-specific databases and which variants may explain either the patient’s phenotype or potentially an unrelated condition.4
Comparison of Whole-genome Sequencing to Whole-exome Sequencing
Although both WGS and WES are clinically available, there are important differences to consider. The largest difference is the amount and content of the data. WGS includes sequence information for all areas covered in the genome, whereas WES is focused on less than 2% of the genome that is known to code for protein and will not report changes in promoter or regulatory regions. With that in mind, you should expect a lot more data with WGS: Approximately, 120 Gb for a 30× WGS compared with approximately 5 to 10 Gb from WES. Although both methodologies can provide greater than 90% of the entire exome sequence, the method by which WES targets or captures only the exon information leads to slightly less coverage of the entire exome as compared with WGS methods.6 However, because of the vastly smaller amount of genomic sequence, throughput and depth of coverage are frequently much greater with WES. In regard to clinical testing, there is not much difference in regard to cost with WES ranging from $4,000 to $15,000 and WGS from $7,500 to $10,000.
Current Limitations
WGS/WES has both technical and clinical challenges. Because of several factors, including alignment programs, short read length, and genome complexity, the ability to use WGS/WES to detect variations larger than a few base pairs is limited, although there has been progress in this area. Currently next-generation sequencing technologies have difficulty accurately calling indels (insertions and deletions), trinucleotide repeats, and copy-number variations. To confidently identify these types of variations, a second testing technology is often required. Thus, interpretation and test reports will be focused on single base-pair, substitution variants. Another challenge is the ability to fully and accurately interpret the resulting sequence information. This is complicated by the currently limited accuracy and completeness of the reference human genome as well as the lack of clinical-grade databases for interpretation.2,4 Ongoing efforts are attempting to address these limitations.
Clinical Whole-Genome Sequencing/Whole-Exome Sequencing Applications
The use of WGS/WES in the clinical setting has already begun, and there is as much excitement surrounding the availability of such testing as there are questions and hesitations. WGS/WES testing has the potential to greatly improve our ability to determine the molecular causation in most Mendelian diseases, and early guidelines for clinical use were recently published by the American College of Medical Genetics and Genomics.7 Researchers have already shown the value of WGS/WES as a tool for identifying candidate genes for genetic conditions with a defined phenotype including Freeman–Sheldon syndrome as well as autosomal dominant retinitis pigmentosa, one of the most genetically heterogeneous Mendelian conditions.6,8
Hereditary cancer syndromes, similar to other Mendelian diseases, have significant genetic heterogeneity, which often necessitates the need to order multiple gene tests. WGS/WES has the potential to enable testing of all possible target genes at once, eliminating the extended time and added cost of sequential gene testing, if needed. This may be particularly helpful in complex disorders such as cancer, where patients may harbor multiple variants that modify their risk. Walsh et al.9 demonstrated this complexity, identifying 2 germline mutations in 3 of 360 ovarian cancer patients. In addition to one germline mutation in either BRCA1 or BRCA2, one of the three participants had a mutation in CHEK2 and the other two had mutations in the MRE11A




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
