- The pathophysiologic mechanisms in type 1 diabetes (T1DM) involve loss of islet β-cell secretory function caused by selective killing of these cells primarily by aggressive autoimmune responses involving both cellular and humoral immune pathways.
- Inflammatory cells heavily infiltrate pancreatic islets leading to insulitis where CD8+ T lymphocytes are thought to be responsible for selective and specific killing of β-cells.
- The complex etiology of T1DM involves a strong genetic predisposition, mainly human leukocyte antigen class II genes, and several putative environmental factors, which are thought to trigger autoimmunity or progression to clinical T1DM.
- A preclinical prodrome in T1DM may vary in duration in which one or more islet autoantibodies may precede insulitis and predict the disease at the early stages of pathologic insult.
- In genetically susceptible individuals with islet autoantibodies, metabolic indicators such as insulin release abnormalities and insulin resistance may best predict T1DM especially near clinical onset.
- Based on the improving understanding of the etiopathogenesis of T1DM, several clinical trials have been launched aiming at halting the autoimmunity responses, retarding disease progression or preserving remaining β-cell function after clinical onset.
Introduction
The differentiation between the two main forms of diabetes mellitus – type 1 (previously known as insulin dependent or juvenile onset) and type 2 diabetes (non-insulin dependent or adult onset) – has been possible for almost 50 years. In 1965, insulitis was rediscovered [1], supporting the view that autoimmune islet inflammation was associated with the etiopathology in type 1 diabetes mellitus (T1DM), a phenomenon absent in type 2 diabetes mellitus (T2DM) [2]. The evidence for islet autoimmunity was further supported during the last decade by the identification of cellular reactivity with islet antigens [3] and the association between T1DM and other organ-specific autoimmune disorders [4]. More importantly, the long sought after antibodies against islet cells (ICA) were finally detected in sera of patients with concomitant T1DM and autoimmune polyendocrine syndrome [5]. At the same time, T1DM was found to be strongly associated with the human leukocyte antigen (HLA) [6]. It was also noted, however, that around 10% of adult patients classified as having T2DM were positive for ICA, a group of patients now commonly known as having latent autoimmune diabetes of adults (LADA) [7]. Several genetic and autoimmune similarities are found between childhood T1DM and LADA; nevertheless, these two entities differ in other genetic and autoimmune processes [8].
T1DM results from an almost complete loss of insulin brought about by selective autoimmune destruction of the pancreatic islets β-cells, which manifests clinically as hyperglycemia-related symptoms and signs. Among non-Hispanic Caucasian populations, more than 90% of T1DM is immune-mediated (also known as T1ADM), in which HLA association is documented and one or more islet cell autoantibodies are detectable at time of diagnosis [2]. The remaining 10%, often termed “idiopathic” (also known as T1BDM as discussed later in this chapter), is highly inheritable but has neither HLA associations nor detectable islet cell autoantibodies [2]. The latter subgroup is found among non-Caucasian ethnic groups such as Asians [9], African-Americans and Hispanic-Americans and is thought to be related to viral infections [2,10]. Clinically, the two forms have similar clinical manifestations and diabetic ketoacidosis may develop in both.
The pathophysiologic mechanisms in T1DM include two distinct stages in genetically susceptible individuals:
1 Triggering of autoimmunity resulting in one or multiple islet cell autoantibodies associated with gradual β-cell killing; and
2 Loss of β-cell secretory function manifested by the loss of first-phase insulin release (FPIR), reduced C peptide levels, then glucose intolerance and finally hyperglycemia (Figure 9.1).
Figure 9.1 Schematic presentation of the natural history of type 1 diabetes (T1DM) showing possible etiopathologic factors and disease markers.
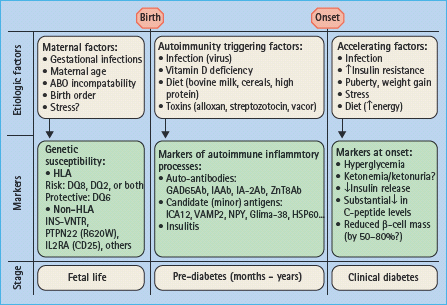
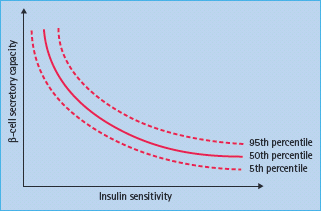
The autoimmune process with mononuclear infiltration of inflammatory cells (insulitis) including autoreactive CD8+ T lymphocytes selectively destroys the β-cells. Both the humoral and cellular pathways of immunity are involved in the disease process; however, the role of B lymphocytes is evident in laboratory animals such as the non-obese diabetic (NOD) mice but not in humans [11]. Islet autoantibodies, however, may be present before insulitis [12] and therefore may not be a direct consequence of insulitis but rather markers of ensuing islet autoimmunity. The induction of islet autoimmunity in genetically susceptible individuals and the appearance of autoantibodies against specific islet cells autoantigens may precede the clinical syndrome by months to several years (Figure 9.2) [13]. During this autoimmunity period the number of islet autoantibodies may reflect how β-cells are gradually destroyed. It is proposed that clinical manifestations became overt after loss of more than 80% of viable β-cell mass [14], although there may be variable degrees of both cellular regeneration and insulin sensitivity (Figure 9.3) [15].
Figure 9.2 Diagrammatic presentation showing the effects of multiple islet autoantibodies on the risk of type 1 diabetes (T1DM) in the Diabetes Prevention Trial Type 1 (DTP-1). Courtesy of Jay Skyler.
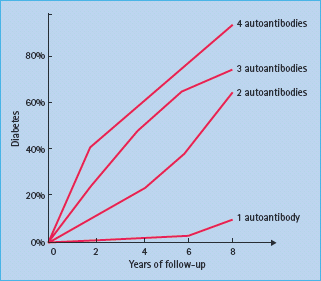
Figure 9.3 The relation between β-cell secretory capacity and insulin sensitivity. Courtesy of Carla Greenbaum, after Kahn et al. [15].
The continuing progress in the understanding of the natural history of T1DM will be dependent on several longitudinal studies aiming at detecting factors that predict the disease and to implement both prevention and intervention trials.
Etiology
The etiology of T1DM is multifaceted and may be divided into genetic and environmental etiology and possible gene–environment interactions. Genetic susceptibility increases predisposition for triggering islet autoimmune responses (Figure 9.1). Genetic factors may also help to accelerate the failure of β-cell secretion in response to exogenous environmental factors such as obesity.
Genetics
The concordance rate of T1DM among monozygotic twins ranges from 30–50% up to 70% [16], depending on follow-up duration, compared to only 10–19% among dizygotic twins [17]. This variability highlights the complexity of etiologic components of T1DM that involves the interaction of multiple genetic factors with a variety of environmental factors. Although more than 85% of T1DM occurs in individuals with no previous first-degree family history, the risk among first-degree relatives is about 15 times higher than the general population [18]. An affected father confers a 6–9% risk of T1DM to his offspring compared to 2–4% if the mother is affected and up to 30% risk if both parents are affected [18,19].
The genetics of T1DM has been studied extensively despite the fact that the mode of inheritance remains uncertain. Recent genome-wide association studies have confirmed the strong associations between T1DM and HLA; however, at least 47 non-HLA genetic factors are thought to contribute. The most prominent genetic factors are listed in Table 9.1.
Table 9.1 The most important genetic factors associated with the risk of type 1 diabetes mellitus (T1 DM).
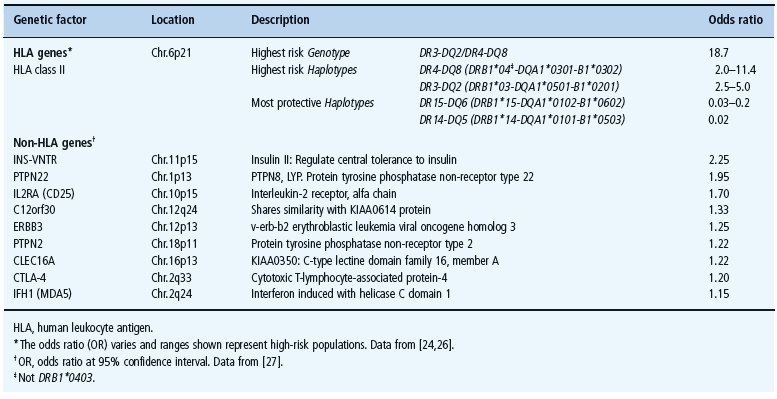
The major histocompatibility complex (MHC) of the short arm of chromosome 6 harbors the main loci involved in the genetic susceptibility of T1DM as well as many other autoimmune diseases [20]. The HLA genes represent almost 50% of the familial risk of T1DM. Certain alleles of the HLA region, such as the HLA class II DR and DQ alleles, are mainly present in specific association with each other, a phenomenon known as linkage disequilibrium. The HLA association of T1DM (Table 9.1) is therefore often described by haplotype or genotype of the individual [21].
The genotype that confers the highest risk of T1DM is the heterozygosity of the two high-risk HLA class II haplotypes: DR3-DQ2 (DRB1*03-DQA1*0501-B1*0201) and DR4-DQ8 (DRB1*04-DQA1*0301-B1*0302) (Table 9.1) [21,22]. One or both of these haplotypes were found in more than 95% of people with T1DM younger than 30 years but also in approximately 40–50% of the general population [23]. The concomitant inheritance of high-risk alleles and haplotypes appears to increase the risk of T1DM significantly through synergistic association of their single risks. For example, in patients with T1DM, DQ8 (DQA1*0301-B1*0302) is mostly inherited with certain variants of DRB1*04 especially DRB1*0401, DRB1*0404, DRB1*0402 but not DRB1*0403 which has negative association (Table 9.1). DQ2 (DQA1*0501-B1*0201), however, is mostly inherited with DRB1 *03 [21,24]. While certain alleles confer higher risk, such as DQB1 *0302, DRB1 *03 and DRB1 *0401, which possesses an independent risk, others confer protection and may “neutralize” high-risk alleles when they are inherited together [25]. The most common protective haplotypes are DQ6 (DQA1*0102-B1*0602 and DQA1*0102-B1*0603), also DQA1*0101-B1*0503 and DQA1*0202-B1*0303 [21,26]. Furthermore, other HLA class II (such as DPB1) and class I alleles have also been associated with T1DM risk and the search for new associations is continuing (for review see [24]).
Using a candidate gene approach, several other non-HLA genes were found to be associated with increased risk of T1DM, but their contribution is less than the HLA haplotype associations (for references see http://www.t1dbase.org/page/Welcome/display or the T1D Genome Consortium website: https://www.t1dgc.org/home.ctm). The most important genes are INS-VNTR on chromosome 11, PTPN22 (LYP) on chromosome 1, IL2RA (CD25) on chromosome 10 (Table 9.1) [27].
Environmental factors
The concordance rate of 50–70% among identical twins [16], the seasonality of diabetes incidence and time of birth [28], the association of diabetes with viral infections [29] and the fact that only 10% of HLA-susceptible individuals develop T1DM [28] are among several observations that indicate a possible etiologic role of environmental factors (Figure 9.1).
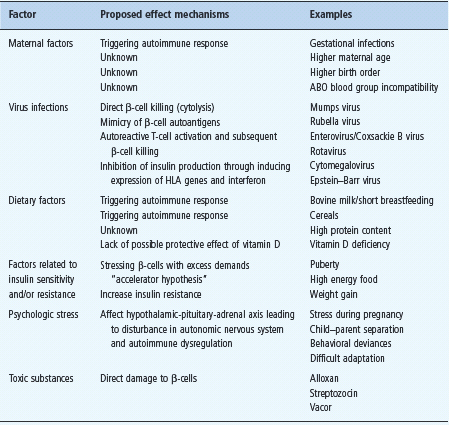
The most prominent environmental factors (Table 9.2) include maternal factors [30], viral infections [29]. dietary [31,32]. high birth weight and growth rate [33]. psychologic stress [34] and toxic substances [35]. The concurrent association of islet autoimmunity and factors increasing insulin resistance such as obesity and accelerated growth may boost the autoimmune destruction of β-cells [36]. The hygiene hypothesis proposes that better sanitation created a pathogen-free environment reducing the exposure to pathogens and their products. According to this hypothesis, the immune systems of children tend to be underdeveloped and therefore prone to autoimmune reactions. Additionally, it was also proposed that younger children received low-level antibodies from their mothers and, when exposed to infections such as enterovirus, it increased their T1DM risk [29].
Table 9.2 The main putative environmental risk factors associated with type 1 diabetes mellitus (T1DM).
Pathogenesis
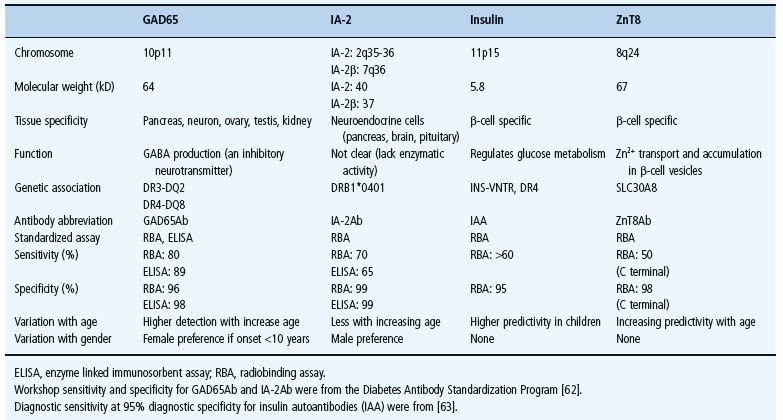
Autoimmune T1DM results from loss of immunologic tolerance to β-cells and environmental factors are thought to be involved in initiation or promotion of autoimmunity or both [28]. The selective destruction of β-cells implies specific mechanisms targeting β-cells by autoimmune reactions, which involve infiltration of pancreatic islets by CD4+ and CD8+ T lymphocytes and macrophages leading to insulitis [1]. During the period preceding the clinical onset, autoantibodies targeting specific islet autoan-tigens such as insulin, glutamic acid decarboxylase (GAD65), islet antigen-2 (IA-2) and zinc transporter (ZnT8) may be detectable for months up to years before hyperglycemia becomes overt [11]. It has been assumed that the occurrence and number of islet autoantibodies such as GAD65Ab and IA-2Ab were associated with insulitis [37]. A recent study, however, detected islet autoantibodies among 62 (4%) individuals out of 1507 pancreatic donors aged 25–60 years [12]. Although those 62 individuals also had HLA susceptibility, only two of them showed insulitis, indicating that the presence of islet autoantibodies is not necessarily a marker of insulitis. The exact role of these autoantibodies is therefore not understood and it needs to be established to what extent they are markers of β-cell destruction by cellular autoimmunity [38].
The intensity and duration of β-cell destruction varies and seems to be related to the presence of high-risk HLA haplotypes especially DR3-DQ2, DR4-DQ8, or both [39]. The HLA class II family molecules are expressed on the surface of antigen-presenting cells (APC) such as dendritic cells and macrophages but also on activated B and T lymphocytes or even activated endothelial cells. High-risk HLA molecules on APC are likely to facilitate activation of CD8+ T lymphocytes by CD4+ T lymphocytes. This activation is indirectly exemplified in T1DM siblings who developed the disease by the age of 12 years because T1DM occurred in 55% of siblings sharing the high-risk HLA DR3-DQ2/DR4-DQ8 genotype compared with only 5% of those who shared zero or one haplotype [40].
It has been widely claimed, based on autopsy studies, that around 80–90% of β-cells are already lost at clinical onset [14,41]. Recent reanalysis of patients who died soon after diagnosis, however, revealed that the level of β-cell loss required for hyper-glycemia was age-dependent, being about 40% in subjects aged 20 years [42]. Additionally, there are suggestions that β-cell regeneration may have taken place, contributing to the approximately 50% of viable β-cells present at diagnosis [43]. The progressive destruction of β-cells is likely to vary in intensity and duration depending on the age at diagnosis [44]. It is a major drawback, however, that direct and precise assessments of β-cell loss before and after diagnosis are not available in humans. Much of the current knowledge of β-cell function prior to the clinical onset has been derived from laboratory animals such as NOD mice and bio-breeding rats [45]. The β-cell destruction in humans may be estimated indirectly by assessing insulin secretion during intravenous glucose tolerance tests (IVGTT). In particular, FPIR measured by insulin or C peptide is thought to reflect loss of β-cells and to predict T1DM [46]. Data from the Diabetes Prevention Trial Type 1 showed that post-challenge C peptide levels were remarkably reduced 6 months before clinical onset [47].
Cellular autoimmunity
The genetic susceptibility of T1DM predisposing to loss of immunologic tolerance and eventual autoimmune killing of β-cells may be explained by a disordered antigen presenting mechanism [43]. The HLA class II molecules are heterodimers that regulate the immune response and are expressed on the surface of APC such as macrophages. The heterodimer binds peptides generated intracellularly either from self-proteins or from exogenous antigens taken up by phagocytosis. The resulting trimolecular complex represents the ligand for the T-cell receptor (TCR). The interaction between the trimolecular complex and the TCR activates the T lymphocyte. The HLA class II molecules on APC are responsible for antigen presentation to T-helper lymphocytes (CD4+). Upon non-antigenic stimulation, macrophages from people with T1DM and the high-risk HLA DQB1*0201/*0302 genotype showed excessive secretion of proinflammatory cytokines and prostaglandin E2 [48]. Cytokines may damage β-cells directly or indirectly by activating other cells such as T and B lymphocytes [49]. The APC presenting β-cell autoantigens may thus be actively involved in the anti-self autoimmune response that may result from failure to sustain self-recognition or from promoting an anti-self response. APC, CD4+ and CD8+ T lymphocytes were all detected in pancreatic autopsies of subjects who died shortly after onset [50], indicating their role in insulitis. Autoreactive CD8+ T lymphocytes may have the most significant role in autoimmune destruction of β-cells [51]. Natural killer cells (NK) may also be found with abnormal activity and count [52]. The detection of autoreactive T lymphocytes in insulitis and in the circulation at the time of diagnosis, in addition to the notion that immunosuppressive drugs such as cyclosporine or anti-CD3 monoclonal antibodies can temporarily abort disease progression, are all considered to support the role of cellular immunity in β-cell destruction [53].
The mechanism involved in β-cell destruction is not yet fully clear. One possible scenario is that β-cells are first destroyed by an environmental factor such as virus. The dying or dead β-cell is next phagocytozed by local dendritic cells (APC), which are then activated and migrate through the lymphatics to a pancreatic draining lymph node. The antigen presentation to and activation of CD4+ T lymphocytes takes place in the lymph node to include activation of CD8+ T lymphocytes specific for islet autoantigens. These islet autoantigen-specific CD8+ T lymphocytes return to the blood circulation, eventually ending up in islets to destroy β-cells. The β-cell killing will generate a new cycle of islet autoantigen presentation known as epitope spreading [49,54]. CD4+CD25+ regulatory T lymphocytes are also thought to have an important role in pathogenesis of T1DM as they may inhibit islet autoantigen specific CD4+ T lymphocytes [54,55]. These cells express FOXp3 from the X chromosome and are important in development of peripheral tolerance.
The identification of islet autoantigen-specific T lymphocytes has been challenging and the development of standardized assays of T lymphocytes specific to islet autoantigens (insulin, GAD65 and IA-2) is still difficult to achieve [53,55]. Soluble HLA class II tetramer assays to assess autoantigen-specific T lymphocytes [56] and ELISPOT (enzyme-linked immunospot) [57] assays to measure cytokines of each T lymphocytes are tests to assess anti-islet autoantigen T-lymphocyte reactivity. During islet autoimmunity prior to the clinical onset these autoantigen-specific T lymphocytes may not be found in peripheral circulation, rather they may accumulate in islets and are therefore hard to detect [58].
Humoral autoimmunity
Islet cell autoantibodies
The identification in 1974 of ICA was achieved using frozen pancreatic sections and indirect immunofluorescence [5]. Four years later, islet surface antibodies (ICSA) were identified [59] and complement-dependent antibody-mediated islet cell cytotoxicity was described in 1980 [60]. Because ICA assays showed wide variations among ICA-positive sera [61]. assays specific to individual autoantigens were later developed to detect autoantibodies against GAD65, IA-2 [62], insulin [63] and recently ZnT8 (Table 9.3) [64]. Islet autoimmunity (single or multiple autoantibodies persistent for 3–6 months) proved to be useful in differentiating T1DM from other forms of diabetes [11]. Multiple islet autoantibodies (≥2) usually appear within 6–12 months following the appearance of the first autoantibody (Figure 9.2) [65]. Nevertheless, some individuals develop transient islet autoantibodies but they are usually solitary and associated with lower risk [66], possibly because of the presence of protective genes such as HLA DR15-DQ6 [25]. One or more of these autoantibodies can be detected months up to years before clinical onset in more than 95% of newly diagnosed patients with T1DM, even as early as in the perinatal period [67]. Moreover, the detection of the four main autoantibodies may predict the disease by as much as 98% (Figure 9.2) [68].
Table 9.3 Characteristics of islet autoantigenes and autoantibodies.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


