Turner syndrome
Historical background
Turner syndrome is a chromosomal disorder due to complete or partial monosomy for the X chromosome, associated with short stature and primary ovarian failure in phenotypic females. The eponym derives from a study published in 1938 by Henry Turner describing seven women with short stature, sexual immaturity, neck webbing, low posterior hairline, and cubitus valgus1(Figure 16-1). Several years earlier, Otto Ullrich had described an 8-year-old girl with short stature, lymphedema of the hands and feet, neck webbing, high arched palate, low-set auricles, and several other features now associated with Turner syndrome.2 Ullrich later recognized that his patient and those of Turner appeared to have the same condition3 and called attention to the work of Bonnevie, who described cervical distention and malformations of the ears, face, and limb buds in mice secondary to dissection of subcutaneous fetal tissues by fluid. Ullrich suggested that fetal lymphatic obstruction may cause neck webbing and other superficial features of Turner syndrome and proposed the eponym Bonnevie-Ullrich to describe this constellation of anomalies. Ullrich’s contributions gave rise to the eponym Ullrich-Turner syndrome sometimes used in Europe.
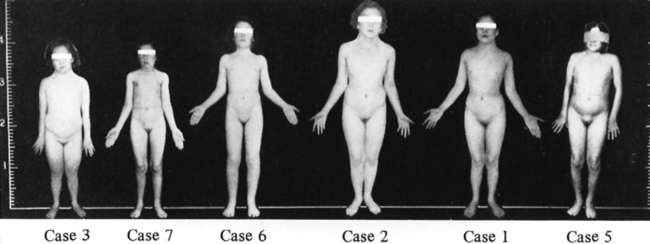
FIGURE 16-1  Patients described by Dr. Henry Turner. Note the height marker at the left indicating the short stature, although with large variation in absolute height among these women. Note also lack of obesity among these women evaluated in the 1930s. (From Turner, H. H. (1938). A syndrome of infantilism, congenital webbed neck and cubitus valgus. Endocrinology, 23, 566.)
Patients described by Dr. Henry Turner. Note the height marker at the left indicating the short stature, although with large variation in absolute height among these women. Note also lack of obesity among these women evaluated in the 1930s. (From Turner, H. H. (1938). A syndrome of infantilism, congenital webbed neck and cubitus valgus. Endocrinology, 23, 566.)
Endocrine and pathology studies of the 1940s revealed primary ovarian failure in women with Turner syndrome, associated with elevated gonadotropins, reduced estrogen and “streak” ovaries consisting of connective tissue depleted of germ cells. These early studies also discovered an extraordinary incidence of hypertension and aortic disease in young women with Turner syndrome.4 The first link between Turner syndrome and sex chromosome anomaly was provided in 1954 by Polani and colleagues, who reported three patients with Turner syndrome and coarctation of the aorta who were sex chromatin negative.5 Soon thereafter advances in cytogenetic identification of specific chromosomes revealed that Turner syndrome was associated with the presence of a single X chromosome (X monosomy).6 These observations were paradigm shifting in our understanding of the role of the human sex chromosomes in sex determination, as reviewed by Opitz and Pallister.7 They also described the significant heterogeneity of patients grouped under the concept of gonadal dysgenesis and pointed out that the terms dysgenesis and agenesis are inaccurate, as fetal ovarian development seems to be normal in Turner syndrome, with degeneration occurring in most cases around the time of birth. Although eponyms have their disadvantages, the designation Turner or Ullrich-Turner syndrome is more specific than gonadal dysgenesis.
Genetics
Chromosomal origins
Refinements in cytogenetic methodology over the last half of the 20th century promoted the elucidation of the chromosomal basis for Turner syndrome and other aneuploidies. The standard cytogenetic karyotype describes the number and morphologic features of condensed metaphase chromosomes under light microscopy (Figure 16-2A). Karyotype studies of the products of conception and newborns have shown that X monosomy is the only chromosomal monosomy compatible with life, as monosomy for the Y chromosome or autosomes have never been reported.8 This distinction among chromosomes is explained by the fact that the Y chromosome has relatively few essential genes aside from those involved in male sex determination and spermatogenesis, and the second X chromosome in females is largely inactivated, or silenced early in development. There are, however, a number of genes that escape X inactivation and that have homologs represented on the Y chromosome.9 Haploinsufficiency for these genes results in short stature and other aspects of the Turner phenotype (discussed later).
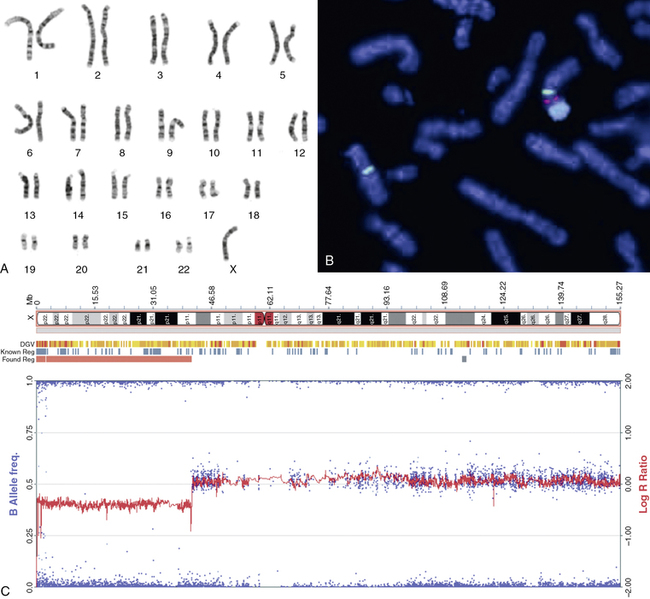
FIGURE 16-2  Sex chromosome analyses. A, Standard G-banded metaphase karyotype in which the 22 pairs of autosomes are grouped according to size and the sex chromosomes placed at the end; in this case, there is only one X chromosome. B, Fluorescent in situ hybridization (FISH) of metaphase chromosomes showing a normal X chromosome on the left side of the micrograph identified by an X centromere-specific probe (DXZ1; green). The abnormal chromosome seen on the right has an X long arm and centromere joined to a Y chromosome centromere (DYZ3; red bifurcated signal) and long arm (DYZ; large blue signal). This is described in cytogenetic terms as der(X)t(X;Y)(p.11.4;p11.2). C, SNP genotyping array of DNA isolated from a Turner patient blood sample. The beta-allele frequency is a measure of the allelic intensity ratio, plotted as (blue) dots with reference to the left y-axis. When there is equal representation of two alternative SNP alleles, the beta allele frequency is 0.5; when only one allele is present, the frequency is 1.0 or 0. The log R ratio is a measure of total signal intensity for the test subject compared to control and is plotted as (red) graph against the y-axis on the right. When test subject signal intensity is equal to that for the control, the ratio is 1:1 and the log is 0. The array results are plotted alongside the X chromosomal ideogram at the top of the panel and show a loss of heterozygosity and reduced signal intensity from chrX: 0-41,500,000, consistent with the deletion of more than half the X chromosome short arm with breakpoint at cytologic band Xp11.4. (B, The FISH micrograph was kindly provided by Dr. Marie-France Portnoi of Service de génétique et embryologie médicales, Hôpital Armand-Trousseau, Paris, France.This image can be viewed in full color online at ExpertConsult)
Sex chromosome analyses. A, Standard G-banded metaphase karyotype in which the 22 pairs of autosomes are grouped according to size and the sex chromosomes placed at the end; in this case, there is only one X chromosome. B, Fluorescent in situ hybridization (FISH) of metaphase chromosomes showing a normal X chromosome on the left side of the micrograph identified by an X centromere-specific probe (DXZ1; green). The abnormal chromosome seen on the right has an X long arm and centromere joined to a Y chromosome centromere (DYZ3; red bifurcated signal) and long arm (DYZ; large blue signal). This is described in cytogenetic terms as der(X)t(X;Y)(p.11.4;p11.2). C, SNP genotyping array of DNA isolated from a Turner patient blood sample. The beta-allele frequency is a measure of the allelic intensity ratio, plotted as (blue) dots with reference to the left y-axis. When there is equal representation of two alternative SNP alleles, the beta allele frequency is 0.5; when only one allele is present, the frequency is 1.0 or 0. The log R ratio is a measure of total signal intensity for the test subject compared to control and is plotted as (red) graph against the y-axis on the right. When test subject signal intensity is equal to that for the control, the ratio is 1:1 and the log is 0. The array results are plotted alongside the X chromosomal ideogram at the top of the panel and show a loss of heterozygosity and reduced signal intensity from chrX: 0-41,500,000, consistent with the deletion of more than half the X chromosome short arm with breakpoint at cytologic band Xp11.4. (B, The FISH micrograph was kindly provided by Dr. Marie-France Portnoi of Service de génétique et embryologie médicales, Hôpital Armand-Trousseau, Paris, France.This image can be viewed in full color online at ExpertConsult) 
The loss or fragmentation of a sex chromosome is most commonly traced to error(s) of recombination and segregation occurring during meiotic divisions.8 The molecular triggers for such errors are poorly understood but appear to differ for autosomes and sex chromosomes.10 Thus, in contrast to trisomy 21, Turner syndrome is not preferentially linked to maternal meiotic errors or age and may be more commonly associated with paternal meiotic errors.11 A less common cause of Turner syndrome is the loss of a sex chromosome due nondisjunction in early embryonic mitotic cell divisions, resulting in a 45,X cell line together with cell lines with normal or supernumerary sex chromosome complement (e.g., 45,X/47,XXX; 45,X/46,XX; 45,X/46,XY).
Epidemiology
Cytogenetic studies found that X chromosome monosomy (45,X) was present in approximately 1/300 spontaneous abortions versus 1/5000 live births,2–13 indicating that most 45,X gestations do not survive to birth. The ratio of 46,X,i(X)q and 46,X,r(X) to 45,X cases increases from early gestation to birth, leading to the view that X monosomy is incompatible with survival and that surviving 45,X girls started out as 46,X, fragmentary X or Y gestations.13 It was hypothesized that fragmentary sex chromosomes are lost due to mitotic instability during the course of fetal development, so the girls appear to be 45,X after birth.13,14 Early in the study of aneuploidy it was thought that monosomy per se would interfere with normal cell proliferation and differentiation.15 However, it has since been learned that mice with pure X monosomy have normal survival, development, and fertility16 and that human 45,X cells are able to proliferate and differentiate into various cell types in vitro.17,18 Thus, it seems that X monosomy need not always be lethal, and the survival and relatively healthy development of some 45,X girls may be related to variations in autosomal genes that compensate for the X chromosome haploinsufficiency. Newborn cytogenetic screening during the 1970s and 1980s found that approximately 1:2500 live-born females had complete or partial X monosomy consistent with Turner syndrome.12,13,19 Danish health registry data indicate, however, that only about half the number of cases predicted by the birth incidence receive a clinical diagnosis.20 This discrepancy may be due to a relatively mild phenotype in some cases. The birth rate for Turner syndrome is going down in some countries,21 associated with the increasing use of fetal ultrasound screening.22
Turner syndrome is reported in all races and countries with similar prevalence. X monosomy does not run in families and is not associated with any known environmental or behavioral factors.23 There is one known case of an apparently pure 45,X woman with spontaneous puberty and pregnancy giving birth to a 45,X daughter.24 Epidemiologic observations including more than 1000 girls with Turner and their families suggest that increased parental age modestly increases the risk of having a child with Turner syndrome.23,25 The single normal X chromosome is identified as maternal in approximately 70% and paternal in origin in ∼30% of cases with cytogenetically confirmed Turner syndrome.26–29 The expected ratio, if each parent has an equal probability of contributing a normal X to offspring, is 2:1. The consistent finding of a slightly greater than expected prevalence of X-maternal cases suggests that there may a greater propensity for meiotic errors involving the sex chromosomes during spermatogenesis. Women with Turner syndrome due to partial Xp deletion or translocation who are fertile may pass on the abnormal X chromosome to offspring.30–32 The possibility of passing on a fragmented sex chromosome underscores the importance of obtaining a full karyotype in the diagnosis of Turner syndrome, because if such a chromosome is found in the child, the parents need genetic testing and counseling about potential recurrence, or transmission if the daughter is fertile. There does not seem to be an increase in monosomy X or any aneuploidy in assisted pregnancies compared to natural conceptions.33 Historical case reports have described an association of trisomy 21 and mosaic X monosomy,34 but this association has not been observed in a large population-based investigation.23
Turner karyotypes
Cytogenetic nomenclature reports the total number of chromosomes followed by the sex chromosomes listed individually, thus the normal female karyotype is “46,XX” and Turner syndrome patients with a single X chromosome are “45,X.” The most common karyotype found in Turner syndrome is 45,X.11,35,36 Mosaicism is defined as the presence of two or more cell lines showing different chromosome constitutions. If more than one cell line is detected, the karyotypes are separated by a slash, with the relative cell counts in brackets. For example, 45,X [10]/46,XX[10] describes an individual with 50:50 mosaicism for X monosomic and normal 46,XX cells based on scoring 20 metaphases. This type of cell line mosaicism occurs due to sex chromosome nondisjunction during postzygotic cell divisions. Depending on the developmental timing and cell populations affected, the clinical phenotype may be attenuated by mosaicism for a normal cell line.
The isoXq chromosome is the most common structural abnormality causing Turner syndrome.11,35,37 It is a mirror image chromosome composed of two copies of the long arm fused head to head with a variable amount of centromeric and proximal Xp sequences in between, most commonly reported as 46,X,i(X)q. Variants that have subtotal Xp deletion are termed isodicentric and pseudo-isodicentric X chromosomes.38,39 Individuals with isoXq chromosome are monosomic for the X short arm and trisomic for the X long arm. The presence of an isoXq chromosome is commonly associated with a 45,X cell line, as the abnormal isoXq chromosome is frequently lost during postzygotic cell divisions. This mosaic chromosomal constitution is reported as 45,X/46,X,i(X)q. Because all cells are monosomic for Xp, there is no tendency for moderation of the monosomic phenotype in this type of cell line mosaicism. The isoXq chromosome is not associated with mosaicism for a normal 46,XX cell line.
A ring X chromosome (rX) is fairly common in the etiology of Turner syndrome; the ring results from fusion of broken ends of short and long arms. Most often the ring has lost a major portion of Xp but retains the large part of Xq, and it is functionally equivalent to Xp deletion. The ring X is commonly lost during cell divisions in the course of development, resulting in mosaicism denoted as 45,X/46X,r(X). A ring X missing the X inactivation site (XIST) at Xq13.2 has been associated with severe mental retardation and somatic features atypical of Turner syndrome.40,41 The XIST locus is required for inactivation of the second X chromosome, and deletion or deficient transcription of XIST may result in failure of X inactivation. The severe phenotype in girls with a small rX or with unbalanced Xp translocation is thought to be due to functional disomy of certain Xp loci that are normally inactivated.42
About 5% of clinically diagnosed Turner syndrome patients have a significant Xp deletion, denoted 46,X,del(X)p, with or without mosaicism for a 45,X cell line. Xq deletions are associated with clinical features of Turner syndrome mainly in patients with 45,X/46,X, del(X)q mosaicism; women with terminal Xq deletion without 45,X cells do not usually have short stature or other features aside from premature ovarian failure.43
Conventional karyotyping reveals mosaicism for 45,X/46,XY or 46,X,fragmentaryY cell lines in about 10% of clinically diagnosed girls with Turner syndrome.11 In cases where the Y chromosome is intact, it is likely that the proportion of normal 46,XY cells was too low in the developing gonad to induce normal testis development and male differentiation. Phenotypic males with 45,X/46,XY chromosomal constitutions may have short stature and congenital cardiovascular malformations similar to those with Turner syndrome44,45 but are considered under the diagnostic category of Disorders of Sexual Differentiation. In girls with Y chromosome structural abnormalities, the sex determining gene SRY may be deleted or inactivated, resulting in absent male differentiation.46 The presence of Y chromosome material is clinically relevant because of the risk for development of a gonadoblastoma in females with Y chromosome material.47 The diagnostic issues related to Y chromosome detection and clinical implications with regard to gonadoblastoma are discussed later in sections on “Diagnostic Tests” and “Gonadoblastoma.”
X chromosome genes and turner syndrome
In a classic article published in 1965, Ferguson-Smith analyzed the karyotypes of 307 patients with various forms of gonadal dysgenesis in relation to their clinical findings.48 He proposed that monosomy for the short arm of the X chromosome was responsible the short stature and congenital malformations found in Turner syndrome, and he noted that Yp deletion was associated with a similar phenotype. Based on this analysis, Ferguson-Smith hypothesized that some Xp genes escape from X-inactivation in 46,XX females and that homologous genes were located on the Y chromosome short arm.48 In addition, he reported that patients with mosaicism for a normal 46,XX line (i.e., 45,X/46,XX) tended to have fewer phenotypic abnormalities.48 These observations were confirmed in subsequent chromosomal banding35 and molecular studies.49
As predicted, genes currently implicated in the Turner phenotype are located on the X chromosome short arm, escape the process of X inactivation, and have functional homologs on the Y chromosome.50 The human sex chromosomes share homologous gene encoding regions located at the termini of the short and long arms. They are termed pseudoautosomal because genes in these regions behave like autosomal genes and do not undergo X inactivation. These regions ensure that X-Y meiotic pairing and recombination takes place, which is essential for male meiosis. The major pseudoautosomal region (PAR1) is located at the Xp and Yp terminal regions and includes at least 25genes.51 Two groups independently identified a PAR1 gene, which when deleted was associated with skeletal deformities and short stature.52,53 This gene is termed SHOX, for “short stature homeobox” and is located ∼500 kb from the telomere of the sex chromosomes at Xp22.3 and at Yp11.3 (Figure 16-3). SHOX encodes a transcription factor that is highly expressed in human bone morphogenetic tissue.54 SHOX haploinsufficiency due to mutation or microdeletion causes Leri-Weill dyschondrosteosis, characterized by mesomelic short stature and shortening and bowing of the radius with a dorsal subluxation of the distal ulna (Madelung deformity).55 Homozygous SHOX deficiency causes Langer mesomelic dysplasia, producing extreme short stature, profound mesomelia, and limb deformity. SHOX mutations or deletions are found in 2% to 15% of children with idiopathic short stature, without obvious skeletal dysmorphology.55,56 These observations have established the view that SHOX haploinsufficiency due to deletion of Xp or Yp terminal regions causes the skeletal anomalies and short stature occurring in Turner syndrome.
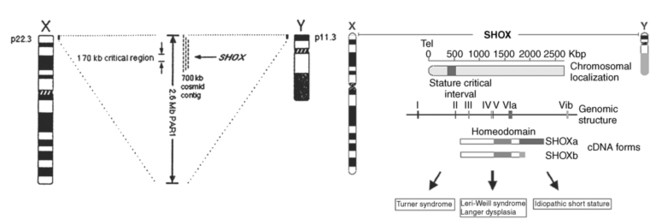
FIGURE 16-3  X and Y chromosome ideograms showing the terminal pseudoautosomal regions (PAR) at Xp22.3 and Yp11.3 where the SHOX gene has been mapped, (A&C) and X chromosome regions historically associated with aspects of phenotype. (From Zinn, A. R. (1997). Growing interest in Turner Syndrome. Nat Genet, 16, 3. Reprinted with permission from Macmillan Publisher Ltd.)
X and Y chromosome ideograms showing the terminal pseudoautosomal regions (PAR) at Xp22.3 and Yp11.3 where the SHOX gene has been mapped, (A&C) and X chromosome regions historically associated with aspects of phenotype. (From Zinn, A. R. (1997). Growing interest in Turner Syndrome. Nat Genet, 16, 3. Reprinted with permission from Macmillan Publisher Ltd.)
Isolated SHOX haploinsufficiency in Leri-Weil or Langer syndromes has not been linked to nonskeletal features of Turner syndrome such as lymphatic obstruction, congenital heart defects, renal anomalies, or hearing loss. It seems likely that haploinsufficiency for other, as yet unknown PAR1 gene(s) contributes to these important defects. This is inferred from the fact that rats and mice that lack PAR1 genes on the sex chromosomes are normal in size, fertile, and without apparent somatic or visceral defects.16 In contrast, canine, feline, and equine species that share a conserved PAR1 region with humans exhibit dwarfism, infertility, and aortic coarctation in X monosomy.50 Several Xp genes outside of PAR1 escape X-inactivation and have Y-homologues, and thus may play a role in Turner syndrome, as recently reviewed.50 Xp deletion mapping data suggested that distinctive neurocognitive characteristics of Turner syndrome are linked to haploinsufficiency for PAR1 or nearby loci.57 Ogata and colleagues ascertained a “lymphedema critical” region at Xp11.4,58 although this localization was not confirmed in a more recent study.59 Bondy and associates have demonstrated that the locus for bicuspid aortic valve and aortic coarctation is telomeric to Xp11.4.60 Hearing loss also maps to Xp deletion.61,62 Genes located on the X chromosome long arm (Xq) are not likely candidates given that girls with normal Xq constitution (e.g., 46,X,del(X)p) and girls that actually have an extra copy of Xq (46,X,i(X)q) have the consistent Turner phenotype.63
X chromosome genomic imprinting
Although haploinsufficiency for Xp genes is clearly implicated in major features of Turner syndrome,64 it is also possible that the exclusive expression of maternally or paternally derived X chromosome may differentiate 45,X girls from the 46,XX population that express an equal ratio of X maternal to X paternal genes secondary to random X inactivation. Important male-female differences such as adult height, brain size, risk for autistic spectrum disorders, abdominal adiposity, and atherosclerosis are independent of gonadal effects and could be influenced by X chromosome genomic imprinting.65 Skuse and colleagues reported impaired social and verbal skills among girls with Turner syndrome who were monosomic for a maternal X chromosome compared to a group monosomic for a paternal X, and they suggested that imprinting of X-linked genes contributes to sex-based differences in risk for autism spectrum disorders.66 Subsequent studies have not supported this view67; however, one MRI study found that brain volumes are greatest in prepubertal girls monosomic for X-maternal, intermediate in 46,XX girls, and smallest in girls monosomic for X-paternal, consistent with a maternal X chromosome dose effect on brain volume.68
Adult height is a sexually dimorphic trait; however, there is no apparent height difference between Turner groups monosomic for either parental chromosome.69–73 Interestingly, adult height in women with Turner syndrome, regardless of X parental origin, tracks maternal but not paternal height.29,70,72–74 One study suggested that response to growth hormone treatment was influenced by the parental origin of the single X chromosome,74 but this has not been confirmed in larger, more recent studies.37,75 Male-pattern abdominal adiposity is observed in females with monosomy for the maternal X chromosome and is associated with an atherogenic lipid profile,76 which may contribute to greater risk for atherosclerosis among women with Turner syndrome and among the general male population, as males are constitutionally monosomic for a maternally derived X chromosome.
Diagnostic tests
A chromosomal karyotype has been required for the definitive diagnosis of Turner syndrome.77,78 This test usually entails obtaining a fresh blood sample from which mononuclear cells are extracted and placed into culture medium. A mitogen such as phytohemagglutinin is used to stimulate lymphocyte proliferation, and after several days, colchicine is used to arrest the cells in metaphase. After fixation, cells are spread on glass slides and treated with Giemsa stain to produce the G-banding pattern, which allows chromosome identification and characterization of major structural anomalies under the light microscope (see Figure 16-2A). Fluorescent-labeled molecular probes corresponding to specific DNA sequences are used to further identify chromosomal deletions or translocations in a process termed fluorescent in situ hybridization or FISH (Figure 16-2B).
For the diagnosis of Turner syndrome, the American College of Medical Genetics recommends scoring a minimum of 20 metaphases from peripheral blood cells.37 If there is a high degree of clinical suspicion but the initial karyotype is normal, another 10 metaphase spreads should be analyzed. In addition, a standard metaphase karyotype may be performed on cultured fibroblasts or interphase FISH may be used on available cells or tissue, although such tests are not well established. In fact, all the clinical and prognostic information guiding standard practice is based on diagnosis using the standard 20-cell karyotype from peripheral blood mononuclear cells. In some cases the standard karyotype reveals small fragments of chromosomal material known as marker or ring chromosomes that cannot be identified as derived from X or Y chromosomes based on morphology. These fragments require identification using FISH with X and Y chromosome specific probes. Some authors suggest further screening for potential Y chromosome sequences using FISH or polymerase chain reaction (PCR) in girls that have a nonmosaic 45,X karyotype.37 This approach has not been widely adopted because of uncertainty about the diagnostic reliability and the unclear clinical significance of “cryptic” Y chromosome sequences.
One study showed that measuring the allelic expression of X chromosome single nucleotide polymorphisms (SNPs) by high throughput sequencing of DNA samples from buccal swabs was able to confirm cytogenetic diagnoses of girls with Turner syndrome.79 The conceptual basis for this approach is that individuals with a single X chromosome will demonstrate lack of heterozygosity for polymorphic X sequences. This method could be used for noninvasive screening of newborns, although the diagnosis would have to be confirmed by standard karyotyping.
A new diagnostic technology relevant to Turner syndrome and other chromosomal disorders uses array-based hybridization to assess both chromosomal sequence copy number and SNP diversity across the entire genome. This new technology provides high resolution virtual, or in silico karyotype analysis (Figure 2C) and is performed on stored blood or tissue samples without need for cell culture. These arrays are able to demonstrate X monosomy, X chromosome deletions, and detect Y chromosome presence equivalent to conventional metaphase karyotypes.80 Their accuracy in detecting low level mosaicism needs further evaluation, however, and additional studies are needed to confirm a correlation between clinical features of Turner syndrome and array-based hybridization diagnoses in a prospective manner.
Indications for karyotype testing
There is a distinct biphasic pattern in the diagnosis of Turner syndrome with a substantial proportion being diagnosed around the time of birth and another large group diagnosed around 12 years of age (Figure 16-4). During infancy, the most common indication for karyotype screening is lymphedema. Residual signs of fetal lymphedema include neck webbing, low hairline, malrotated, low-set ears, and drooping eyelids. Swelling or puffiness of the hands and feet accompanies peripheral lymphedema, which is common in infants with Turner syndrome. Skeletal features and growth delay may be present but difficult to appreciate during the first few years. Another important indication for karyotype analysis is the presence of congenital cardiovascular malformation, especially aortic coarctation and aortic valve disease in a female infant or young girl.
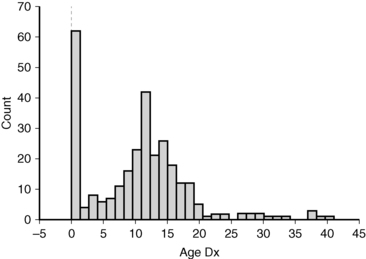
FIGURE 16-4  Age of Turner diagnosis. The histogram shows the age of diagnosis for 292 patients evaluated in the NIH Turner natural history protocol between 2005 and 2010. The median age of diagnosis was 11 years.
Age of Turner diagnosis. The histogram shows the age of diagnosis for 292 patients evaluated in the NIH Turner natural history protocol between 2005 and 2010. The median age of diagnosis was 11 years.
A karyotype should be considered in the evaluation of girls with short stature, especially if associated with typical skeletal features such as micrognathia, high-arched palate, short metacarpals, or cubitus valgus as described later (skeletal anomalies and growth failure). Girls and young women presenting with delayed or absent puberty associated with elevation of gonadotropins should have a karyotype analysis. Finally, Turner syndrome is not infrequently discovered by karyotype analysis in women with infertility or premature menopause, as illustrated by the late age of diagnosis for 7% of the National Institutes of Health (NIH) cohort (see Figure 16-4).
Differential diagnosis
The standard karyotype analysis plays an essential role in differential diagnosis of Turner syndrome. Noonan described male and female children with neck webbing, short stature, and congenital heart disease,81 and in the past boys with this disorder were classified as “male Turner syndrome.” Noonan syndrome is a genetically heterogeneous autosomal dominant disorder with no sex predominance and is associated with a normal karyotype. The Noonan congenital heart defects include pulmonic valve stenosis and hypertrophic cardiomyopathy81—in contrast to the Turner cardiovascular phenotype that includes mainly left ventricular outflow tract defects. Puberty may be delayed, but girls are usually fertile whereas boys may have cryptorchidism. The nomenclature, evaluation, genetic counseling, and endocrine management that are appropriate to the girl with Turner syndrome do not apply to patients with Noonan syndrome. Another consideration in the differential diagnosis of a girl undergoing evaluation for short stature with some skeletal features associated with Turner syndrome and similarly affected siblings is SHOX mutation/deletion or Leri-Weill syndrome. Girls with delayed or absent puberty, normal height, and 46,XX or 46,XY karyotypes are thought to have isolated mutations/deletions of genes involved in gonadal differentiation. These patients were historically grouped with girls with Turner syndrome under the category “gonadal dysgenesis,” but they clearly have very different genetic, medical, and psychosocial issues.
Prenatal diagnosis
The prognosis for prenatally detected Turner syndrome depends in large measure on the indications for the testing. When an abnormal fetal ultrasound showing cystic hygroma or fetal hydrops leads to genetic testing and demonstration of nonmosaic 45,X fetal karyotype, there is virtual certainty of a clinically significant diagnosis of Turner syndrome and a high risk of fetal demise.82 A characteristic ultrasound of a 14-week-old Turner fetus with a cystic hygroma is shown in Figure 16-5. In contrast, an incidental prenatal diagnosis of Turner syndrome with a normal fetal ultrasound is often associated with a mild postnatal phenotype.83 The postnatal outcomes for fetuses mosaic for 45,X and normal 46,XX or 46,XY cell lines are quite variable and correlate poorly with phenotypes of children ascertained on clinical grounds with Turner syndrome or mixed gonadal dysgenesis.84 Over 90% of 45,X/46,XY cases ascertained by amniocentesis or villous chorionic sampling appear to be normally developed males at birth.85 The outcome for fetuses with 45,X/46,XX karyotypes appears to be similarly mild.83,86 Given these observations, it is important that prenatal counseling inform families that 45,X/46,XY and 45,X/46,XX gestations with normal fetal ultrasound may present a milder phenotype than that of patients diagnosed clinically. Moreover, even the 45,X fetus with cystic hygroma may be viable and enjoy a good quality of life. Importantly, prenatal diagnoses of sex chromosome anomaly should be reevaluated postnatally with a standard peripheral blood karyotype in all cases.
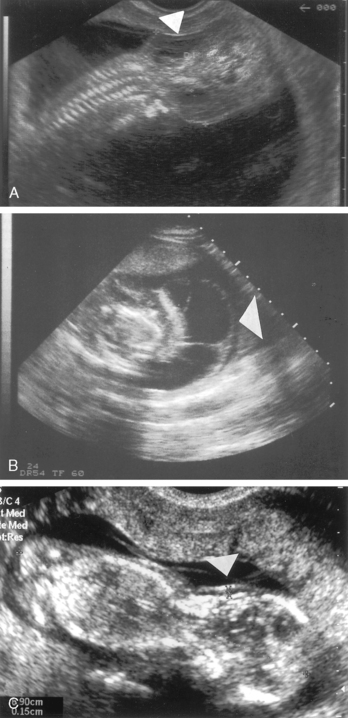
FIGURE 16-5  A, A 14-week-old fetus with Turner syndrome and a cystic hygroma (arrow). B, A 13-week-old fetus with a normal karyotype and normal nuchal translucency of 1.5 mm (arrow). C, Same fetus as in view A in transverse plane. Large septated cystic hygroma (arrow) can be seen around the fetal neck. (Courtesy of Pekka Taipale, MD, PhD, Kuppio University Hospital, Finland.)
A, A 14-week-old fetus with Turner syndrome and a cystic hygroma (arrow). B, A 13-week-old fetus with a normal karyotype and normal nuchal translucency of 1.5 mm (arrow). C, Same fetus as in view A in transverse plane. Large septated cystic hygroma (arrow) can be seen around the fetal neck. (Courtesy of Pekka Taipale, MD, PhD, Kuppio University Hospital, Finland.)
As genomic technology advances, we are seeing new approaches to prenatal screening for genetic abnormalities. Massively parallel genomic sequencing is able to characterize small amounts of cell free fetal DNA in the maternal blood stream by 10 weeks’ gestation, and this approach may eventually supplant the use of the maternal “analyte” screen. However, suspicions of aneuploidy based on cell free fetal DNA, maternal analytes or fetal ultrasound still require direct confirmation of the chromosomal complement from in situ fetal tissue obtained from amniocentesis or chorionic sampling. More advances on the genomic technology front may replace the traditional karyotype of fetal tissue. Comparative genomic hybridization (CGH) arrays are equal to traditional karyotype in identification of trisomy 21 and sex chromosome anomalies, including 45X from fetal samples obtained by amniocentesis or chorion samples.87 CGH is more sensitive to microdeletions or duplications compared to traditional karyotype analysis, but it may not detect balanced translocations or cell line mosaicism associated with Turner syndrome, for example, 45,X/47,XXX. The clinical outcomes of pregnancies identified by the new genomic technologies have not yet been studied.
Phenotypic features
Since the original description of Turner, it has been recognized that there are a multiplicity of findings in patients with Turner syndrome and that phenotypic features vary remarkably between patients with the same karyotype (Figure 16-6). Table 16-1 summarizes the most common features determined by physical diagnosis and standard medical screening.

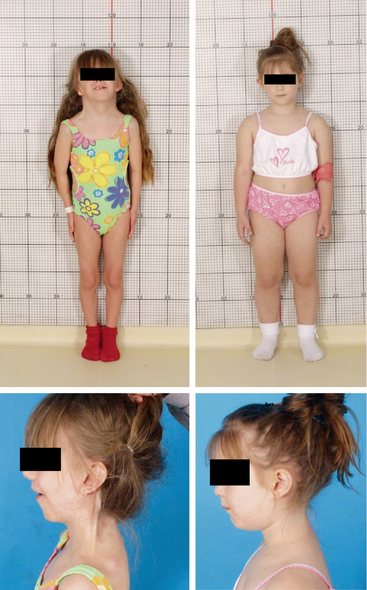
FIGURE 16-6  Phenotypic variability in Turner syndrome. Both of these 7-year-old girls with short stature have Turner syndrome with a 45,X karyotype confirmed in analysis of 50 lymphocytes. The girl on the left was diagnosed at birth due to prominent neck webbing and low-set and posteriorly rotated ears. She also has micrognathia and a low posterior hairline. In contrast, the girl on the right was diagnosed at age 7 due to short stature without “classical” stigmata of Turner syndrome, and she is more typical of the clinical presentation of the majority of girls with Turner syndrome diagnosed in the 21st century. (This image can be viewed in full color online at ExpertConsult)
Phenotypic variability in Turner syndrome. Both of these 7-year-old girls with short stature have Turner syndrome with a 45,X karyotype confirmed in analysis of 50 lymphocytes. The girl on the left was diagnosed at birth due to prominent neck webbing and low-set and posteriorly rotated ears. She also has micrognathia and a low posterior hairline. In contrast, the girl on the right was diagnosed at age 7 due to short stature without “classical” stigmata of Turner syndrome, and she is more typical of the clinical presentation of the majority of girls with Turner syndrome diagnosed in the 21st century. (This image can be viewed in full color online at ExpertConsult)
Lymphatic obstruction
The appearance of the 45,X fetus illustrated in Figure 16-7 dramatically shows the fetal edema that occurs in many conceptuses with Turner syndrome. The edema appears to result from lymphatic malformations and obstruction.88 The cystic hygromas are due to delayed formation of communications between the jugular and central lymphatics draining into the heart that normally develop between the fifth and sixth weeks of gestation. Peripheral lymphatic hypoplasia or aplasia has also been demonstrated using lymphangiography in adult patients with Turner syndrome.89

FIGURE 16-7  A 45,X abortus demonstrating generalized lymphedema. Note the distended cervical region. With resolution of the edema, the redundant skin may cicatrize, resulting in a webbed neck. The edema of the hands and feet may persist and be present at birth. (From Gellis, S. S., & Feingold, M. (1978). Picture of the month. Am J Dis Child, 132, 417. Copyright © 1978, American Medical Association.)
A 45,X abortus demonstrating generalized lymphedema. Note the distended cervical region. With resolution of the edema, the redundant skin may cicatrize, resulting in a webbed neck. The edema of the hands and feet may persist and be present at birth. (From Gellis, S. S., & Feingold, M. (1978). Picture of the month. Am J Dis Child, 132, 417. Copyright © 1978, American Medical Association.)
Webbed neck, or pterygium colli, is the most obvious consequence of nuchal lymphatic obstruction and this is seen in approximately 25% of patients diagnosed currently. The tenting of developing scalp and facial structures is believed to cause low-set, rotated auricular structures, down-sloping eyes, drooping eyelids, and low posterior hairline (see Figure 16-6). It has been suggested that this mechanical distention during fetal development may be responsible for lush hair growth, including eyelashes and eyebrows. Edema of the dorsum of the hands and feet at birth signals peripheral lymphedema, which usually resolves, although some patients demonstrate chronic involvement. Others may complain of intermittent or recurrent edema, often after the institution of estrogen replacement therapy.
Skeletal anomalies and short stature
The most common physical abnormality associated with Turner syndrome is short stature. The impairment is most pronounced along the longitudinal body axis. This gives affected individuals the visual appearance of being stocky or squarely shaped and accounts for the predominantly illusory finding of widely spaced nipples.90 The short neck is secondary to hypoplasia of one or more of the cervical vertebrae.91 Long bone growth is selectively impaired, resulting in relatively short legs and an increased upper-to-lower segment ratio.92 Individual bones are affected to varying degrees. For example, cubitus valgus is commonly appreciated and can be measured as the angle of intersection of the long axis of the upper arm with the long axis of the supinated forearm when the elbow is fully extended. Normally, in adult women this angle is approximately 12 degrees—whereas in adult men it is approximately 6 degrees.93 The major determinant of the angle is the depth of the inner lip of the trochlea of the ulna relative to the outer lip. In many patients with Turner syndrome the angle is between 15 and 30 degrees (Figure 16-8) as a consequence of developmental abnormalities of the trochlear head.
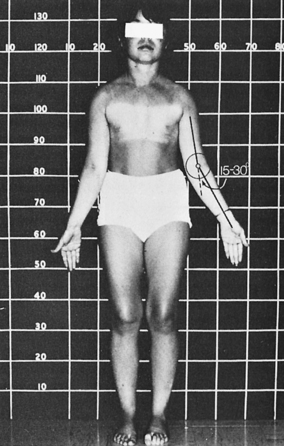
FIGURE 16-8  A 16-year-old girl with Turner syndrome and absence of puberty. Note absence of most characteristic stigmata save short stature and an increased carrying angle (cubitus valgus).
A 16-year-old girl with Turner syndrome and absence of puberty. Note absence of most characteristic stigmata save short stature and an increased carrying angle (cubitus valgus).
A shortened fourth metacarpal is visible on a bone age radiograph (Figure 16-9A) and is visible on exam by depression of the knuckle when the patient makes a fist. The Madelung deformity is found in 7% to 8% of patients and is caused by bowing of the radius coupled with dorsal subluxation of the distal ulna (Figure 16-10).94 This anomaly also occurs as part of the Leri-Weill syndrome.
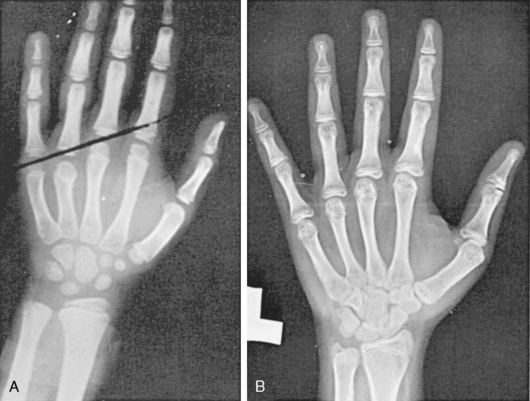
FIGURE 16-9  Two characteristic hand radiographs. A, Short fourth metacarpal, the tip falling below a straight line drawn between the third and fifth metacarpals. B, Generalized lacy (“fish net”) appearance of the carpals and tufting of the distal phalanges, characteristic of the osteoporotic appearance of the bones of patients with Turner syndrome.
Two characteristic hand radiographs. A, Short fourth metacarpal, the tip falling below a straight line drawn between the third and fifth metacarpals. B, Generalized lacy (“fish net”) appearance of the carpals and tufting of the distal phalanges, characteristic of the osteoporotic appearance of the bones of patients with Turner syndrome.

FIGURE 16-10  A 19-year-old-patient with Turner syndrome and bilateral bayonet-like Madelung deformities of the wrists.
A 19-year-old-patient with Turner syndrome and bilateral bayonet-like Madelung deformities of the wrists.
Spinal scoliosis or kyphosis is reported in a significant number of patients95 and may be secondary to vertebral anomalies or leg-length inequality. Micrognathia or receding chin and high arched palate are common and thought to be due to disturbances of the development of facial bones.
Bones of the hand and wrist on bone age radiographs often demonstrate osteopenia,96 associated with ballooning of the tips of the terminal phalanges.97 Both findings are illustrated in Figure 16-9B.This osteoporotic appearance is observed in childhood, suggesting that it may be more related to the developmental role of SHOX than to primary estrogen deficiency. In support of this view, cortical bone mineralization is selectively reduced in girls and women with Turner syndrome, independent of estrogen exposure.98 Long bone fractures associated with minimal trauma appear to occur more frequently among girls with Turner syndrome.99
Short stature is the most common phenotypic feature of Turner syndrome. The first comprehensive assessment of skeletal growth deficits was reported by Ranke and colleagues in 1983.100 Patterns of growth are illustrated in Figure 16-11, which shows cross-sectional height and velocity data from a series of 150 Turner children who had not received therapy to promote growth. The observations were supplemented by Davenport and coworkers,101 who distinguished stages of growth deficit: mild intrauterine growth retardation with average birth length 1 standard deviation (SD) below the mean; a period of mild growth deceleration from birth to age 3.102 After age 3, there is continued deceleration, so that between ages 3 and 13 years Turner syndrome girls fall farther and farther away from the normal height curves and, if untreated, fail to experience a pubertal growth spurt but continue to grow at a slow rate for several more years.
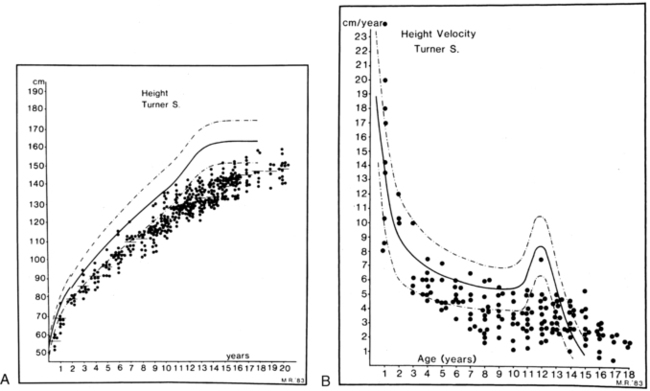
FIGURE 16-11  Height and height velocity in Turner syndrome. A, Three hundred eighty-four single measurements of height for 150 children with Turner syndrome. B, Height velocity from a total of 159 measurements. The normal ranges are shown by the heavy and dashed lines. (From Tanner, J. M., Whitehouse, R. H., & Takaishi, M. (1965). Standards from birth to maturity for height, weight, height velocity and weight velocity: British children. Arch Dis Child, 41, 454, 613; Ranke, M. D., Pfluger, H., Rosendahl, W., et al. [1983]. Turner syndrome: spontaneous growth in 150 cases and review of the literature. Eur J Paediatr, 141, 81.)
Height and height velocity in Turner syndrome. A, Three hundred eighty-four single measurements of height for 150 children with Turner syndrome. B, Height velocity from a total of 159 measurements. The normal ranges are shown by the heavy and dashed lines. (From Tanner, J. M., Whitehouse, R. H., & Takaishi, M. (1965). Standards from birth to maturity for height, weight, height velocity and weight velocity: British children. Arch Dis Child, 41, 454, 613; Ranke, M. D., Pfluger, H., Rosendahl, W., et al. [1983]. Turner syndrome: spontaneous growth in 150 cases and review of the literature. Eur J Paediatr, 141, 81.)
A positive correlation is found among height at diagnosis,103 ultimate height achieved, and midparental height.104 The final height deficit, using comparative data on adult heights in patients with different ethnic backgrounds, is approximately 20 cm.105 Lyon and associates106 used the data from Ranke and colleagues and from three other European centers to synthesize a series of growth curves for Turner syndrome. The curves provided mean height and SD values for age and determined a mean adult height 143.1 cm. Lyon and colleagues also noted a strong correlation between the initial height on these Turner curves and the adult height essentially independent of bone age at the time of the first height. Thus, they concluded that one could project the adult height of a girl with Turner syndrome based on her height at an earlier age. The deficit in longitudinal bone growth in Turner syndrome has been attributed to the deleterious effect of SHOX haploinsufficiency, and a similar deficit is seen in isolated SHOX defects such as Leri-Weill syndrome. Growth hormone deficiency is not implicated in Turner short stature.
Ovarian insufficiency
The earliest studies of ovarian pathology in women with Turner syndrome described fibrous streaks devoid of oocytes and follicles, and thus initially it seemed that gonads failed to develop or were “dysgenetic.” Subsequent studies revealed that early stages of ovarian development appeared normal, with expected numbers of oocytes and primordial follicles at 14 to 16 weeks of gestation in Turner fetuses.107 However, at later stages of development, Turner ovaries were relatively depleted of oocytes and had few developing follicles, suggesting an accelerated rate of oocyte demise and follicular atresia,108,109 although follicles in various stages of development are detected in some teenage girls with Turner syndrome.110 The cause for the high rate of oocyte attrition in most girls with Turner syndrome is unknown. It has been suggested that aneuploidy contributes to oocyte demise due to meiotic mishaps.111 Another possible explanation for oocyte loss is that diploid expression of unknown X chromosome gene(s) is required for normal oocyte generation or survival. The chromosomal location of putative fertility genes has remained elusive, with gonadal failure frequently observed in Xp deletions with intact Xq complement and in some cases of Xq deletion with normal Xp complement.112 Terminal Xq deletions have been associated with premature ovarian failure in women who have few if any features of Turner syndrome.113
Ovarian function and potential for spontaneous puberty and even pregnancy are variable and sometimes difficult to assess among girls with Turner syndrome. An important Swedish study using ovarian biopsy has shown that a karyotype with mosaicism for 45,X and 46,XX cell lines is the most significant positive predictive factor for the presence of ovarian follicles, whereas karyotypes indicating nonmosaic 45,X or structural defects of one X chromosome were significant negative correlates for the presence of follicles.114 Clinical factors such as normal follicle stimulating (FSH) and anti-Mullerian hormone (AMH) levels and spontaneous start of puberty were also significant positive predictors of follicle presence, although less robust than the blood karyotype.114 According to a large, multicenter Italian study that included more than 500 girls with Turner syndrome, spontaneous puberty occurs in about 15% of those with pure 45,X and in 30% of girls with a second cell line with more than one X chromosome (i.e., 45X/46XX; 45X,47XXX).36 Puberty may fail to progress to menarche in some girls, and menarche may be followed by oligomenorrhea or anovulatory cycles in others, so that the actual percentage of young women that maintain normal menstrual cycles by age 20 is less than 5%.
Spontaneous pregnancy occurs in 2% to 3% of women with Turner syndrome.36,115,116 This is more common among women with mosaicism for 46,XX or 47,XXX cell lines, but there are several well-documented reports of spontaneous pregnancies in 45,X women who had no evidence of mosaicism despite intensive investigation.24,116,117 An early case series reported a high frequency of fetal mortality or malformation for spontaneous pregnancies among women with Turner syndrome.118 However, this has not been observed in more recent, population-based studies116,119 nor in women with X monosomy.116,120 If the mother has Turner syndrome due to a sex chromosome structural abnormality, the abnormal chromosome may be passed on to her offspring. The risk of maternal complications with spontaneous or assisted pregnancies is very high for women with Turner syndrome. These concerns are discussed at the end of this chapter in the “Reproductive Options” section.
Gonadoblastoma
The gonadoblastoma is a benign tumor that consists of large germ cells surrounded by small cells with variable granulosa, lutein, or Sertoli-like morphology. This type of tumor strongly resembles the histology of a developing gonad, hence the appellation “gonadoblastoma.” This proliferative island of tissue within the dysgenetic gonad has a potential for steroid production and for malignant transformation into dysgerminoma.47 The gonadoblastoma is found in approximately 40% of females with 46,XY mixed gonadal dysgenesis; the frequency among girls with Turner syndrome and Y chromosomal material is between 10% to 30%.47 These frequency data are based on a histologic examination of ovaries that were “prophylactically” removed from girls with Y chromosome material in their karyotype. There are no studies showing morbidity or mortality related to gonadoblastoma in Turner syndrome. Analyses of Danish health registry data did not find increased morbidity or mortality related to any type of ovarian tumor among women with Turner syndrome.20,121 A large British registry study ascertained gonadoblastoma diagnoses in 8% of women with Y chromosome in their peripheral blood karyotype, but it did not report any clinical data associated with the diagnosis.122
Prophylactic gonadectomy has been standard practice since the time that an excess of gonadal tumors was first appreciated in females with Y chromosomes and intra-abdominal gonads. Supporting this approach was the view that the gonads in these cases were nonfunctional, essentially contributing nothing to the patient but risk. However, we have learned that spontaneous puberty and even pregnancy may occur in individuals with Turner syndrome and Y chromosome material.32,123,124 For example, Portnoi and colleagues reported the case of a girl diagnosed with Turner syndrome at age 8 because of short stature who was found to have translocation of Y chromosome material onto the X chromosome (see Figure 16-2B). The patient’s family declined the advice for castration and the girl went on to develop spontaneous puberty and eventually had a well-supervised successful pregnancy.32 All the clinical experience with gonadoblastoma/dysgerminoma in girls with Turner syndrome derives from cases where the karyotype included visible Y chromosome material or the patient had clinical evidence of virilization, hence the recommendation for gonadectomy was applied only to individuals with visible Y chromosome material or virilization.125,126 With the advent of molecular amplification technology, several studies have reported detection of Y chromosome sequences in 45,X girls without evidence of Y chromosome material on their karyotype, sometimes with a histologic demonstration of gonadoblastoma after gonadectomy. Some authors suggest that all 45,X patients undergo molecular screening for cryptic Y chromosome material. On the other hand, PCR amplification may yield false positive results127 and immature ovaries may contain nests of germinal cells resembling the benign gonadoblastoma, so the risk of overtreatment is a real concern.
Cardiovascular system
Spectrum and etiology of congenital cardiovascular malformations
Congenital cardiovascular malformations (CVM) are the most serious, life-threatening consequences of X chromosome monosomy.20,122,128 Coarctation of the aorta in Turner syndrome was documented many years ago5,48,129; however, early series reporting the spectrum of CVM in Turner syndrome included patients with Noonan syndrome,130 which has a different cardiovascular phenotype.81 The frequency and spectrum of Turner-specific CVM was established in more recent studies using chromosomal karyotyping and cardiovascular imaging (Table 16-2). Obvious CVMs occur in approximately 75% of spontaneously aborted Turner fetuses and 30% of living patients. Obstructive lesions of the left ventricular outflow tract predominate, ranging in severity from nonstenotic bicuspid aortic valve to aortic stenosis, coarctation of the aorta, aortic aneurysm, and mitral valve anomalies (see Table 16-2). The most severe form of left-sided hypoplasia (hypoplastic left heart syndrome) also occurs in Turner syndrome and has a very poor prognosis.131,132 The association of Turner syndrome and left-sided cardiovascular malformation is distinctive among malformation syndromes.
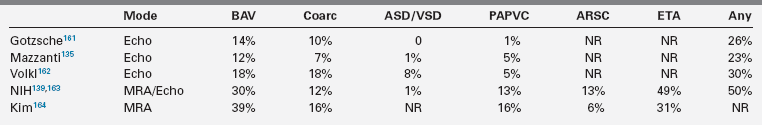
Clark described a significant association between neck webbing and aortic coarctation in girls with Turner syndrome.133 This association was supported by observations on aborted fetuses134 and additional clinical studies reporting a significantly higher prevalence of aortic coarctation and bicuspid aortic valve (BAV) in girls with neck webbing.135,136 Clark proposed that engorged fetal lymphatic channels might compress the ascending aorta and alter intracardiac blood flow, resulting in the spectrum of left ventricular outflow tract (LVOT) defects.133 However, in a study of 45,X fetuses with severe aortic arch defects and aortic valve abnormality, Miyabara and coworkers described a generalized hypoplasia of fourth branchial arch tissues, which in their view was more consistent with defective migration of neural crest cells, as opposed to mechanical effects due to lymphectasia.137 Moreover, LVOT defects occur in Turner patients without neck webbing, and neck webbing is found in some girls with normal cardiovascular structure and function demonstrated by advanced imaging,138 which seems inconsistent with a cause-and-effect relationship. It may be more likely that haploinsufficiency for sex chromosome gene(s) causes central fetal lymphedema and aortic heart defects independent of each other, with penetrance conditioned by autosomal genetic variation and possibly environmental effects.
Interestingly, nonsyndromic LVOT defects such as aortic coarctation and BAV in the general population are not associated with lymphedema or neck webbing and are more common in males than in females by a ratio of approximately 3:1. The cardiovascular phenotype is found in patients with Turner syndrome with deletion of just the short arm of the X or Y chromosome,48,139 suggesting that loci important for LVOT development are present on Xp and Yp. The putative candidate gene would escape X inactivation in females and be expressed from both the X and Y chromosomes in males. The Yp allele may be more prone to disruption in males as a consequence of inherent Y chromosome mutability.140 PAR1 localization of an LVOT related gene(s) may explain the greater prevalence of these defects among males, as the meiotic recombination rate for this region is sevenfold greater in males than females,141 increasing risk for Yp gene disruption.
Aortic complications
Complications of congenital cardiovascular disease are the leading cause of morbidity and premature mortality in Turner syndrome.128,142,143 The major cardiovascular complications include aortic valve disease and aortic dilation, dissection, or rupture. The aortic valve is congenitally abnormal in approximately 30% of life-born girls with Turner syndrome, but it is not detected in many individuals until the third decade of life or later due to inadequate screening.139,144 Among 74 patients with bicuspid aortic valve studied at the NIH (median age 30 year, range 7 to 67), 55% had normal aortic valve function, 30% had mild aortic regurgitation, and 15% had moderate to severe aortic regurgitation.139 Aortic stenosis was present in only 2/74 patients with BAV in this series. BAV prevalence was equal in pediatric and adults groups, but the likelihood of valve dysfunction was higher among adults. Interestingly, the large NIH screening study utilizing advanced imaging confirmed an early small study by Miller and coworkers indicating BAV in 34% of girls with Turner syndrome.145 It is clearly important to screen for aortic valve pathology in all girls and women with Turner syndrome, because the valve may deteriorate over time, leading to heart failure. Moreover, the presence of aortic valve abnormality is linked to aortic pathology with increased risk for aortic dilation, dissection, and rupture.146–153
The presence of an abnormal aortic valve is associated with relative dilation of the ascending aorta, using age and body surface area nomograms, in girls and women with Turner syndrome.139,151 The degree of dilation is greatest in patients with abnormal aortic valve function139,151 but may be present in individuals with normal valve function as well. Moreover, mild-moderate ascending aortic dilation is found in approximately 10% of Turner patients who have a normal tricuspid aortic valve and normal blood pressure.139,151 There is some evidence for a generalized vasculopathy in adults with Turner syndrome with enlarged diameter of carotid and brachial arteries154 and aneurysms of other arteries.155 The term aortopathy has come into wider use to describe aortic disease of diverse etiologies including Marfan syndrome and related vascular disorders that are not associated with valvular disease, in addition to aortic disease associated with BAV. It is unknown at present whether the risk for aortic complications in Turner syndrome is more similar to the former or the latter. This is a critical issue, because indications for intervention, the most effective type of surgery, and therapeutic responses to pharmacologic treatment are likely to be different in these categories.
The risk for serious aortic complications is increased in girls and women with Turner syndrome by 100-fold or greater compared to the general female population, estimated from an epidemiologic study of Danish registry data.150

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


