Neonatal diabetes mellitus
Definition
Neonatal diabetes mellitus (NDM) was strictly defined as severe hyperglycemia occurring in the first month of life, lasting at least 2 weeks, and requiring insulin therapy to control blood glucose.1 These strict criteria have been progressively loosened, initially to include onset in the first 6 months of life, when autoimmune type 1 diabetes mellitus (T1DM) is highly unlikely, thereby implicating a genetic cause for pancreatic malformation, or faulty insulin synthesis and secretion. Increasingly, it has become recognized that, depending on the degree of expression and the inherent capacity of the genetic mutation to disrupt normal insulin output, several of the genetic forms can initially present with symptoms up to 9 months to 1 year, or even later.2–4 Indeed, pedigree analyses conclusively demonstrate that the same defect that causes permanent or transient NDM can be present in parents or other first-degree relatives and be diagnosed as T1DM, monogenic diabetes of youth (MODY), or type 2 diabetes mellitus (T2DM) as subsequently detailed.5,6 Herein lies the importance of understanding the genetic basis of NDM, for although considered rare, with a reported incidence varying from approximately 1 in 100,000 to 1 in 400,000 live births,7 these entities have taught us much about the genetic pathways involved in the formation of the exocrine and endocrine pancreas.8,9 For example, it has been shown that the specific combination of three transcription factors, Ngn3, Pdx1 and Mafa, known to be implicated in the determination of cell lineage during pancreas formation, can reprogram adult mouse exocrine pancreatic cells into cells that closely resemble pancreatic β cells.10 Such information is essential for the ultimate ability to generate β cells and whole islets as potential therapies for T1DM.11 Equally important is the demonstration that activating mutations in the pore-forming Kir6.2 and its regulatory subunit SUR1 of the KATP-regulated potassium channel, which keep the channel open and hence limit or preclude insulin secretion resulting in NDM (Figure 9-1), can be overcome by high-dose sulfonylurea therapy, which restores endogenous insulin secretion in response to feeding.12 Due to the restoration of endogenous insulin secretion together with an incretin effect in response to feeding as opposed to intravenous glucose, this oral treatment provides better metabolic control than multiple daily injections of insulin or insulin pumps and with a better quality of life.7,12 These findings emphasize the benefits of research for understanding pathophysiology and choosing appropriate treatment. Indeed, for those whose NDM is caused by mutations in the KATP channel that respond to sulfonylureas, these treatments border on the miraculous.
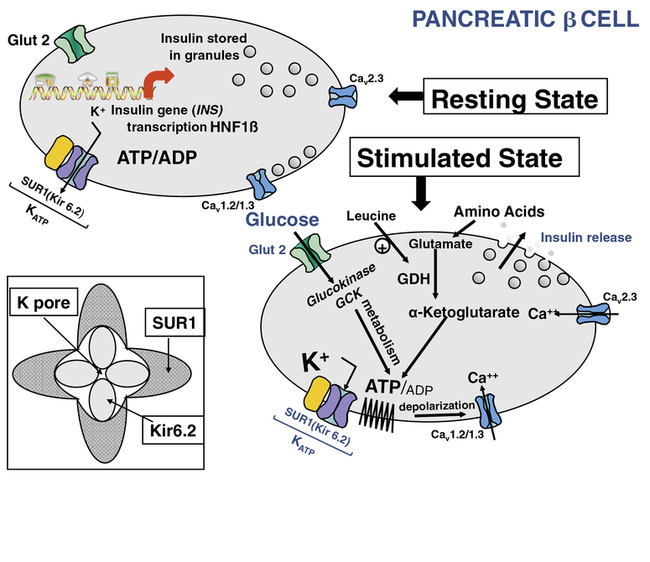
FIGURE 9-1  This is a schematic representation of the role of KATP channels in nutrient regulation of insulin secretion. In the resting (nonfed) state, depicted in the upper left panel, insulin synthesis and storage are regulated by transcription from the insulin gene (INS) and by transcription factors such as hepatic nuclear factor 1β (HNF1β); mutations in INS or HNF1β as well as other genes can cause transient or permanent neonatal diabetes mellitus (NDM).The KATP channel is composed of four subunits of the inward rectifying potassium channel 6.2 (Kir 6.2) encoded by the KCNJ11 gene on chromosome #11) and four regulatory subunits of sulfonylurea receptor 1 (SUR1), encoded by the ABCC8 gene, also located on chromosome #11 (inset lower left). In the fasting nonfed state, the KATP channel remains open. However, in the stimulated (fed) state (panel lower right) glucose concentration increases and enters the β cell in a concentration-dependent, but insulin-independent manor via the Glut 2 glucose transporter encoded by the gene SLCA2A. Glucokinase (GCK) phosphorylates glucose to G6P and its metabolism generates ATP. The resultant change in ATP:ADP causes closure of the KATP channel, accumulation of intracellular potassium, membrane depolarization leading to opening of voltage-gated calcium channels and secretion of stored insulin, as depicted in the lower right panel. Metabolism of amino acids such as glutamate also generates ATP, which stimulates insulin secretion as described for glucose. The amino acid leucine acts as an allosteric stimulus to glutamate dehydrogenase (GDH), which enables metabolism and generation of ATP. Activating mutations of the KATP channel maintain it in an open state to varying degrees, in spite of ATP generation, therefore preventing insulin secretion, which leads to diabetes mellitus, including NDM. Inactivating mutations in KATP genes prevent normal channel opening, maintaining varying degrees of channel closure, and hence constant insulin secretion that causes HI (see Chapter 21). ATP, adenosine triphosphate; ADP, adenosine diphosphate; Ca++ -, calcium; GDH, glutamate dehydrogenase; KATP, ATP-regulated potassium channel; INS, insulin gene; HNF1β, hepatic nuclear factor1β; SUR1, sulfonylurea receptor1, K+, potassium; Kir6.2, potassium inward rectifying channel family6 subtype2; GCK, glucokinase, Glut2, glucose transporter 2. (This image can be viewed in full color online at ExpertConsult)
This is a schematic representation of the role of KATP channels in nutrient regulation of insulin secretion. In the resting (nonfed) state, depicted in the upper left panel, insulin synthesis and storage are regulated by transcription from the insulin gene (INS) and by transcription factors such as hepatic nuclear factor 1β (HNF1β); mutations in INS or HNF1β as well as other genes can cause transient or permanent neonatal diabetes mellitus (NDM).The KATP channel is composed of four subunits of the inward rectifying potassium channel 6.2 (Kir 6.2) encoded by the KCNJ11 gene on chromosome #11) and four regulatory subunits of sulfonylurea receptor 1 (SUR1), encoded by the ABCC8 gene, also located on chromosome #11 (inset lower left). In the fasting nonfed state, the KATP channel remains open. However, in the stimulated (fed) state (panel lower right) glucose concentration increases and enters the β cell in a concentration-dependent, but insulin-independent manor via the Glut 2 glucose transporter encoded by the gene SLCA2A. Glucokinase (GCK) phosphorylates glucose to G6P and its metabolism generates ATP. The resultant change in ATP:ADP causes closure of the KATP channel, accumulation of intracellular potassium, membrane depolarization leading to opening of voltage-gated calcium channels and secretion of stored insulin, as depicted in the lower right panel. Metabolism of amino acids such as glutamate also generates ATP, which stimulates insulin secretion as described for glucose. The amino acid leucine acts as an allosteric stimulus to glutamate dehydrogenase (GDH), which enables metabolism and generation of ATP. Activating mutations of the KATP channel maintain it in an open state to varying degrees, in spite of ATP generation, therefore preventing insulin secretion, which leads to diabetes mellitus, including NDM. Inactivating mutations in KATP genes prevent normal channel opening, maintaining varying degrees of channel closure, and hence constant insulin secretion that causes HI (see Chapter 21). ATP, adenosine triphosphate; ADP, adenosine diphosphate; Ca++ -, calcium; GDH, glutamate dehydrogenase; KATP, ATP-regulated potassium channel; INS, insulin gene; HNF1β, hepatic nuclear factor1β; SUR1, sulfonylurea receptor1, K+, potassium; Kir6.2, potassium inward rectifying channel family6 subtype2; GCK, glucokinase, Glut2, glucose transporter 2. (This image can be viewed in full color online at ExpertConsult)
Incidence
Early estimates placed the incidence at approximately 1 in 500,000,1 and most authorities still cite figures in the 1 in 200,000 to1 in 400,000 range.13 As increased awareness has led to these entities being recognized more frequently, the reported incidence has risen considerably. In populations with high rates of consanguinity, it has been reported that 1 in 21,000 births is associated with NDM.14 A large representative database for pediatric diabetes reported that the incidence of NDM represented approximately 1 case in 89,000 live births in Germany, and a similar incidence occurred in Italy.15,16 However, in three other European countries, the incidence was reported to be 1 in 260,000 live births.17 In the SEARCH for Diabetes in Youth Study involving 15,829 subjects aged < 20 years diagnosed with diabetes during the years 2001-2008, 39 were diagnosed before the age of 6 months.18 Among these 39 subjects with onset < 6 months of age, 35 had permanent neonatal diabetes and an additional 3 had transient NDM leaving 1 subject whose status remained unknown.18 Hence, the total prevalence was approximately 0.246% or approximately 1 in 4000 of all children diagnosed with diabetes in that study during that time frame. The majority were classified by their primary care providers as having T1DM and treated with insulin; only seven underwent mutational analysis for three of the most common genes (KCNJ11, ABCC8, and the insulin gene INS) and five of these seven had mutations in one of these three genes.18
Clinical presentation
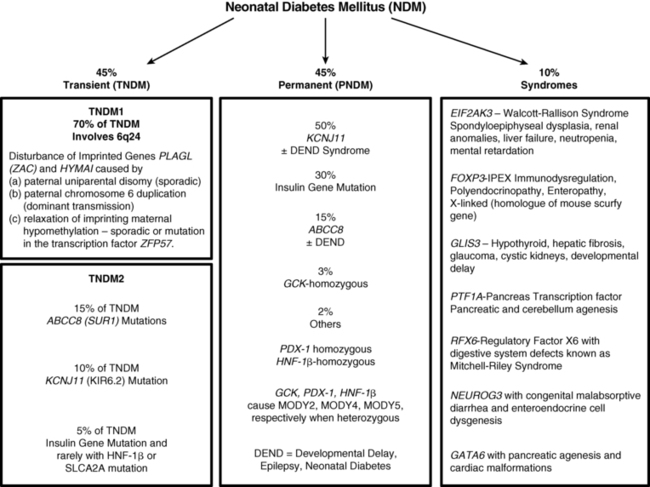
Typically, infants affected with NDM present in the first days to weeks of life with intrauterine growth retardation, reflecting the in utero deficiency of insulin and emphasizing the role of insulin as a determinant of fetal growth.19 Their small size and low birth weight markedly contrasts with the large birth weight and size of infants with inactivating mutations in the same genes that lead to hyperinsulinemic hypoglycemia (see Chapter 21). A disproportionate number are born prematurely at < 37 weeks’ gestation.20 Hyperglycemia leads to osmotic diuresis and avid feeding from breast or bottle despite which the infants fail to thrive. Delay in diagnosis from not considering the possibility of diabetes mellitus in a newborn may lead to severe dehydration and life-threatening diabetic ketoacidosis. Some have congenital malformations including macroglossia and umbilical hernia, reminiscent of Beckwith-Wiedemann syndrome; these usually represent inheritance of genes with disordered methylation as a result of uniparental (paternal) disomy, paternal inherited duplications, or maternal hypomethylation of a differentially methylated region (DMR) at chromosome 6q24 (Figure 9-2).20 Some of these infants have dysmorphic facies as well as renal tract anomalies such as hydronephrosis and vesicoureteral reflux, a variety of cardiac anomalies, hypothyroidism, and hand-finger anomalies.20 A coarse facial appearance together with epilepsy and later manifestations of developmental delay constitute the developmental delay, epilepsy, neonatal diabetes (DEND) syndrome associated with severe mutations of the KCNJ11 gene.12 However, clinical manifestations involving organs other than the pancreas may be part of the syndrome of a variety of genetic mutations associated with NDM as listed in Figure 9-3.
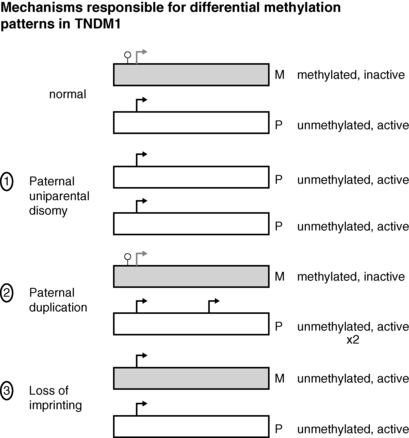
FIGURE 9-2  Transient neonatal diabetes mellitus type 1 is caused by differential expression of PLAGL1 and HYMAI, genes on chromosome 6q24 that result from differential expression of the maternal and paternal alleles. Normally the maternal allele remains methylated and inactive, whereas the paternal allele is unmethylated and active. The differential expression of these genes can occur by one of three mechanisms: (1) paternal uniparental disomy, where both genes are of paternal origin; (2) paternal duplication, so that two active paternal genes are expressed; (3) loss of imprinting, also referred to as “relaxation of imprinting,” on the maternal genes on 6q24 as illustrated here. See text for details.
Transient neonatal diabetes mellitus type 1 is caused by differential expression of PLAGL1 and HYMAI, genes on chromosome 6q24 that result from differential expression of the maternal and paternal alleles. Normally the maternal allele remains methylated and inactive, whereas the paternal allele is unmethylated and active. The differential expression of these genes can occur by one of three mechanisms: (1) paternal uniparental disomy, where both genes are of paternal origin; (2) paternal duplication, so that two active paternal genes are expressed; (3) loss of imprinting, also referred to as “relaxation of imprinting,” on the maternal genes on 6q24 as illustrated here. See text for details.

Classification
The entities of NDM are conveniently divided into transient and permanent, which together make up approximately 90% of the genetically recognized types of NDM; a third category constituting approximately 10% of known entities is associated with mutations that affect organs other than the pancreas and hence present as syndromes whose spectrum of clinical and radiologic features provide clues to the cause (see Figure 9-3). Transient NDM is so named because hyperglycemia is transient, and in the most common form, named TNDM1, it resolves at a median age of 3 months, although rarely it has taken up to 48 months for resolution.20 Generally, those that do not remit by the end of the first 6 months to 1 year are considered to have permanent neonatal diabetes. Transient neonatal diabetes predominantly involves disturbances in the imprinted genes pleomorphic adenoma gene-like 1 (PLAGL-1) also known as zinc finger gene regulating apoptosis and the cell cycle (ZAC) and hydatidiform mole-associated and imprinted transcript (HYMAI) on the differentially methylated region (DMR) of chromosome 6q24. This entity has been named transient neonatal diabetes mellitus type 1 (TNDM1) and comprises the majority, estimated at some 70% of all forms of TNDM. A second form of TNDM, named TNDM2 to distinguish it from the defects in the 6q24 region, includes mutations in the ATP-regulated potassium channel involving predominantly mutations in ABCC8 (SUR1) and KCNJ11 (KIR6.2), with a small minority being due to recessive insulin gene mutations, mutations in the transcription factor HNF1β, and mutations in SLCA2A, the gene encoding the GLUT2 transporter, that together account for ∼30% of TNDM (see Figure 9-3).
Transient neonatal diabetes mellitus (TNDM)
TNDM1
TNDM1 is caused by the overexpression of the paternal PLAGL-1, which is a proapoptotic zinc finger protein, and HYMAI gene, encoding an untranslated mRNA, which arise as a result of uniparental disomy (UPD), duplication of chromosome region 6q24, or relaxation of imprinting of the maternal methylated genes on chromosome 6 (see Figures 9-2 and 9-3). It is important to emphasize that these chromosomal changes cause altered expression of genes, rather than representing mutations. For example, PLAGL-1 (ZAC) has antiproliferative properties and is thought to function as a tumor growth suppressor expressed only on the paternal allele21; overexpression in fetal life is believed to lead to underdevelopment of the pancreas. Although the precise mechanisms by which overexpression of PLAGL-1 lead to TNDM1 are not known, overexpression of ZAC in a clonal pancreatic beta cell line impairs glucose-stimulated insulin translation and secretion.22 A transgenic mouse model expressing the human TNDM1 locus (6q24) is characterized by impaired glucose homeostasis with hyperglycemia in the neonatal period and impaired glucose tolerance with reduced insulin responses to IV glucose as adults.23 The pancreata of these animals display reduced expression of endocrine differentiation factors, notably Pdx-1, Ngn3, and Pax6. There is also a reduction in the number of insulin staining cells and reduced insulin content or insulin secretion despite normal or elevated beta cell mass at all postnatal periods.23,24 Thus, this model recapitulates TNDM1 and suggests that altered expression of ZAC/HYMAI cause impaired development of the endocrine pancreas as well as impaired beta cell function.23,24 This mouse model also demonstrates resolution of abnormal insulin secretion with restoration of normal glucose tolerance during the “juvenile” phase of mouse development between 1.5 and 2 months of life, during which there is an approximate doubling of beta cell number that compensates for the reduced insulin synthesis and secretion of each cell. Also, as in humans, the compensatory increase in beta cell mass is not sustained resulting in a mild diabetes mellitus characterized by normal fasting glucose but hyperglycemia after glucose challenge.23,24 Overall, despite the recapitulation of the key features of the human disease, the mouse model displays milder features.23,24 One possible reason for this milder phenotype in mice is that pancreatic expression of the mouse ortholog Zac-1 declines drastically during gestation and early postnatal growth in mice, whereas expression of the ZAC gene in human pancreas declines between the second trimester and adult life.25 More important, ZAC was specifically expressed only in the islets of the human fetus, whereas Zac-1 was predominantly expressed in mesenchyme of the mouse embryo which may explain the milder features in the mouse model of TNDM1.25
In patients with TNDM1 due to hypomethylation of the maternal DMR of chromosome 6q24, there may be hypomethylation of other maternally imprinted loci (HIL) throughout the genome.26 In cases that display a more generalized hypomethylation of imprinted loci (HIL), the majority have a mutation in the transcription factor zinc finger protein 57 (ZFP57).27 HIL also occurs in the Beckwith-Wiedemann syndrome and this likely explains the macroglossia, umbilical hernia, and several congenital abnormalities described in TNDM1.28 In a large multinational cohort involving 163 patients with TNDM1,20 the authors describe intrauterine growth retardation with a mean birth weight of 2001 ± 417 g (mean ± SD ) and adjusted Z score for birth weight of −2.5. The mean age of presentation was 8±12 days with a median of 4 days and a mode of 1 day. Mean gestation was 37.8 ± 2.4 weeks and prematurity was significantly more common than in the general population.20 Remission occurred at a mean age of 4.5 ± 5.8 months with a median at 3 months. Age at presentation was positively correlated to gestational age, but age at remission was negatively correlated with adjusted birth weight. Thus, the higher the birth weight, the earlier the remission and vice versa. This would be consistent with the effects of insulin on intrauterine growth, so that the larger infants would have the milder defect and therefore tend to enter remission sooner. Congenital anomalies were significantly more frequent in patients with UPD of chromosome 6 or hypomethylation of multiple imprinted loci. Hypomethylation defects were overrepresented in patients born after assisted conception. Thus, babies with TNDM1 generally present with diabetes mellitus within the first days of life, are small, and may have been born prematurely. The presence of congenital anomaly suggests UPD or multiple HIL, and among the latter, almost one in seven conceived with assisted reproductive techniques. Macroglossia is present in about 50%, umbilical hernia in ∼25%, and facial dysmorphism in about 20%. Cardiac and renal anomalies (∼9%), hand abnormalities (∼8%), and hypothyroidism (∼4%) also may be present.20 Remission, when it occurs, is usually around 3 months and about half of these patients will revert to varying degrees of hyperglycemia in the teen years or later.20 An unusual manifestation is hypoglycemia with hyperinsulinemia, following remission of diabetes in patients with 6q24 methylation defects, most often parental unidisomy.29 Modern molecular techniques permit diagnosis to be established, and these may influence treatment.
Ideally, these patients should be treated with an insulin pump using insulin diluted to 1:10 with appropriate diluent in order to provide the lower required amounts (0.2 to 1 u/kg/day).30 Patients with TNDM1 are sensitive to insulin and respond with rapid and remarkable catch-up growth after several weeks of the treatment. Progressive reduction of the insulin dose required to control blood glucose while avoiding hypoglycemia heralds the onset of remission.
TNDM-2
Transient neonatal diabetes mellitus type 2 is a convenient way to classify conditions associated with a neonatal form of diabetes mellitus that remits during infancy and may recur later in life, but is due to mutations in genes regulating insulin secretion rather than expression of imprinted genes.31,32 The majority of these entities are due to activating mutations in the ABCC8 and KCNJ11 genes, which, respectively, code for the SUR1 and Kir6.2 subunits of the KATP channel (see Figure 9-1).31,32 Recessive, loss of function mutations in the insulin gene itself may occasionally be responsible for TNDM233; the autosomal dominant insulin gene mutations are associated with permanent NDM.2,31,32 There are rare reports of HNF1β34 and SLC2A2 mutations35 also associated with TNDM.
The normal state of the KATP channel is to remain open; insulin secretion occurs when the channel closes in response to an increase in ATP generated from the metabolism of glucose or amino acids, thereby changing the ATP:ADP ratio. Closure of the channel with intracellular retention of the K+ causes depolarization of the plasma membrane, opening of voltage-gated calcium channels, influx of calcium, and secretion of insulin. Activating mutations in ABCC8 or KCNJ11 alter the ability of the channel to respond to the change in the ATP:ADP, so that the channel remains open to some extent, efflux of K+ from the β cell continues, permitting the cell membrane to remain hyperpolarized and therefore resulting in decreased insulin secretion. These same mechanisms also are responsible for the most common form of permanent neonatal diabetes mellitus as subsequently discussed.31,32 It remains unclear how or why remission occurs, but it has been shown in vitro that mutations causing TNDM have a less pronounced effect on channel function compared with mutations that cause PNDM.36 The ability of ATP to close the channel in vitro correlates with the severity of neonatal diabetes mellitus, including the severe permanent form associated with the DEND syndrome, which demonstrates the greatest resistance to closure by ATP in vitro.36,37 Both resistance to closure due to activating mutations, or resistance to opening of the channel due to inactivating mutations, segregate with certain mutations as illustrated in Figure 9-4, adapted from reference 36. As shown in the figure, near the fulcrum of this spectrum, those with minor defects may be prone to either develop milder type 2 diabetes, or be resistant to the development of diabetes by virtue of enhanced insulin secretion (see Figure 9-4).
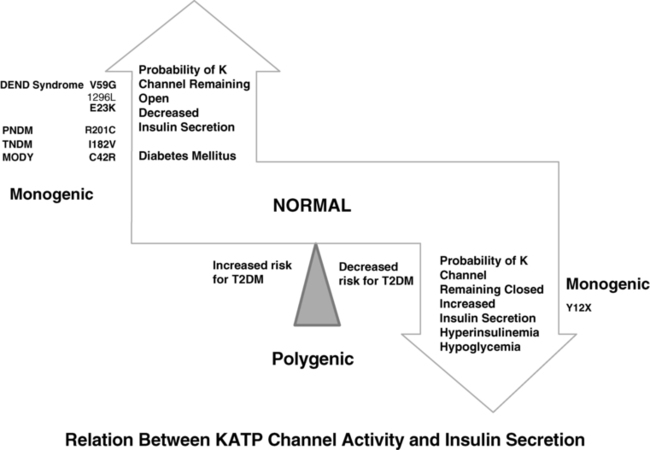
FIGURE 9-4  This is a schematic representation of the relationship between KATP channel activity and insulin secretion. Activating mutations of KCNJ 11 gene maintain the channel in an open state and hence limit insulin secretion. With progressively increasing probability of the potassium channel remaining open, the severity of the diabetes increases from a mild increased risk for T2DM, to monogenic diabetes of youth (MODY), transient neonatal diabetes mellitus (TNDM), permanent neonatal diabetes mellitus (PNDM), and in the most severe state the DEND syndrome (developmental delay, epilepsy, neonatal diabetes) as illustrated on the left. By contrast, mutations that increase the probability that the channel remains closed also increase the likelihood of persistent insulin secretion and lead to hyperinsulinemia and hypoglycemia as illustrated on the panel on the right; in milder forms they may decrease the risk for T2DM by maintaining a higher insulin secretion. Common genetic defects are illustrated in each as examples. (Modified from Ashcroft FM [2005]. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest 115:2047-2058. Figure 6.)
This is a schematic representation of the relationship between KATP channel activity and insulin secretion. Activating mutations of KCNJ 11 gene maintain the channel in an open state and hence limit insulin secretion. With progressively increasing probability of the potassium channel remaining open, the severity of the diabetes increases from a mild increased risk for T2DM, to monogenic diabetes of youth (MODY), transient neonatal diabetes mellitus (TNDM), permanent neonatal diabetes mellitus (PNDM), and in the most severe state the DEND syndrome (developmental delay, epilepsy, neonatal diabetes) as illustrated on the left. By contrast, mutations that increase the probability that the channel remains closed also increase the likelihood of persistent insulin secretion and lead to hyperinsulinemia and hypoglycemia as illustrated on the panel on the right; in milder forms they may decrease the risk for T2DM by maintaining a higher insulin secretion. Common genetic defects are illustrated in each as examples. (Modified from Ashcroft FM [2005]. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest 115:2047-2058. Figure 6.)
In comparison to TNDM1, patients with TNDM2 generally have greater birth weight, present or are diagnosed with diabetes mellitus later, remit later, and recur earlier (Table 9-1). Family members of patients with these forms of TNDM2 may have diabetes that was diagnosed in adulthood as T1DM, T2DM, or MODY and yet harbor the same heterozygous mutations as the proband with NDM.4–6 This reflects the variable penetrance of these genes or upstream factors that may modify the expression of the gene in different individuals. Confirming the presence of an ABCC8 or KCNJ11 mutation in a case of NDM is important for management because most of these KATP channel mutations respond to sulfonylurea treatment both at the time of initial diagnosis or later at the time of relapse.12,31 During their remission phase, these patients do not require therapy.
TABLE 9-1
Comparison of Clinical Characteristics of Patients with KATP Channel Mutations to Patients with 6q24 TNDM (data given in median [range])

From Flanagan, S. (2013). Transient neonatal diabetes. Diapedia, Rev. 21. Retrieved from www.diapedia.org/other-types-of-diabetes-mellitus/41040851198/transient-neonatal-diabetes.
A few patients have been reported with recessive loss of function mutation in the INS gene in patients with TNDM.33 These patients appear to enter remission at a median age of 12 weeks; insulin is required before remission and after relapse, which has occasionally been reported.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


