Adrenal cortex and its disorders
HISTORY, EMBRYOLOGY, AND ANATOMY
PLASMA STEROIDS AND THEIR DISPOSAL
CLINICAL AND LABORATORY EVALUATION OF ADRENAL FUNCTION
GENETIC LESIONS IN STEROIDOGENESIS
Congenital Lipoid Adrenal Hyperplasia
Disorders Resembling Lipoid CAH: P450scc Deficiency and SF1 Deficiency
3β-Hydroxysteroid Dehydrogenase Deficiency
17α-Hydroxylase/17,20-Lyase Deficiency
P450 Oxidoreductase Deficiency
Lesions in Isozymes of P450c11
GLUCOCORTICOID THERAPY AND WITHDRAWAL
Commonly Used Glucocorticoid Preparations
Withdrawal of Glucocorticoid Therapy
Stress Doses of Glucocorticoids
Copyright © 2014 by Walter L. Miller and Christa E. Fluck. Published by Elsevier Inc. All rights reserved.
History, embryology, and anatomy
History
The history of adrenal research has been reviewed.1 The adrenal glands apparently were first described in 1563 by the Italian anatomist Bartolomeo Eustaccio, better known for his description of the eustachian tube of the ear. Medical interest in the adrenals as something other than an anatomic curiosity began in the mid-19th century with Addison’s classic description of adrenal insufficiency and Brown-Sequard’s experimental creation of similar disorders in animals subjected to adrenalectomy. The signs and symptoms of glucocorticoid excess due to adrenal tumors were well known by 1932, when Cushing described the pituitary tumors that cause what is now known as Cushing disease. Effects of adrenalectomy on salt and water metabolism were reported in 1927, and by the late 1930s, Selye had proposed the terms glucocorticoid and mineralocorticoid to distinguish the two broad categories of actions of adrenal extracts.
Numerous adrenal steroids were painstakingly isolated and their structures determined during the 1930s in the laboratories of Reichstein and Kendall, leading to their sharing the 1950 Nobel Prize in medicine. Many of these steroids were synthesized chemically, providing pure material for experimental purposes. The observation in 1949 that glucocorticoids ameliorated the symptoms of rheumatoid arthritis greatly stimulated interest in synthesizing new pharmacologically active analogs of naturally occurring steroids. The structures of the various adrenal steroids suggested precursor/product relationships, leading in 1950 to the first treatment of congenital adrenal hyperplasia with cortisone by both Wilkins and Bartter. This opened a vigorous era of clinical investigation of the pathways of steroidogenesis in a variety of inherited adrenal and gonadal disorders. The association of cytochrome P450 with 21-hydroxylation was made in 1965, and some of the steroidogenic enzymes were then isolated in the 1970s, but it was not until the genes for most of these enzymes were cloned in the 1980s that it became clear which proteins participated in which steroidal transformations.2 The identification of these genes (Table 13-1) then led to an understanding of the genetic lesions causing heritable disorders of steroidogenesis. At the same time, studies of steroid hormone action led to the discovery of steroid hormone receptors in the 1960s, but it was not until they were cloned in the 1980s that their biology has begun to be understood.3
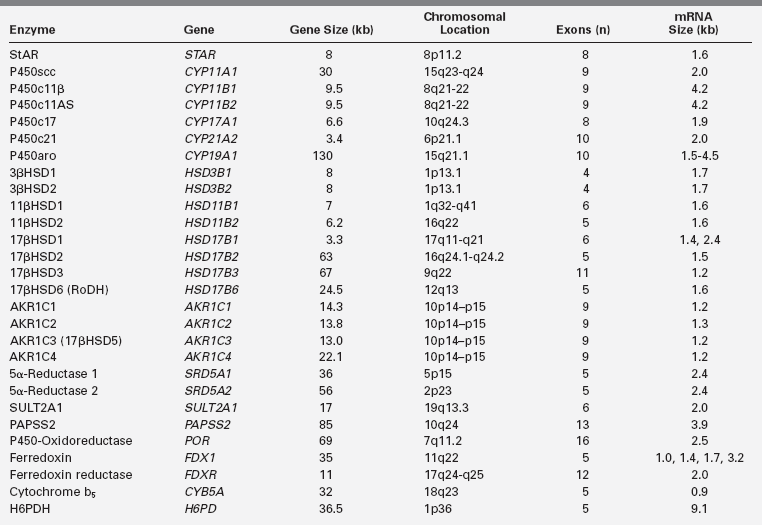
Embryology
The cells of the adrenal cortex are of mesodermal origin, in contrast to cells of the adrenal medulla, which are derived from the neuroectoderm. In human embryos, adrenogonadal progenitor cells first appear at around the fourth week of gestation as a thickening of the coelomic epithelium (or intermediate mesoderm) between the urogenital ridge and dorsal mesentery4 (Figure 13-1). These progenitor cells give rise to the steroidogenic cells of the gonads and to the adrenal cortex. The adrenal and gonadal cells then separate, with the adrenal cells migrating retroperitoneally to the cranial pole of the mesonephros and the gonadal cells migrating caudally. Between the seventh and eighth week of development, the adrenal primordium is invaded by sympathetic cells derived from the neural crest that give rise to the adrenal medulla. By the end of the eighth week, the rudimentary adrenal has become encapsulated and is clearly associated with the upper pole of the kidney, which at this time is much smaller than the adrenal.5
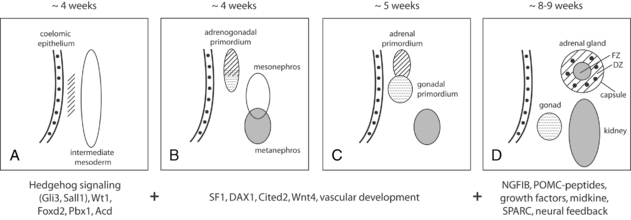
FIGURE 13-1  Overview of human adrenal development. A-C, The adrenogonadal primordium develops at around 4 weeks’ gestation, after which the adrenal primordium becomes a distinct structure that then migrates retroperitoneally to the cranial pole of the mesonephros. D, By 8 to 9 weeks’ gestation, the adrenal gland is encapsulated, contains chromaffin cells (black), and has distinct fetal (FZ) and definitive zones (DZ). Some of the signaling molecules, transcription factors and growth factors implicated in adrenal development are shown here, although the exact timing and interaction of many of these factors remains poorly understood at present. (Adapted with permission from Else, T., & Hammer, G. D. (2005). Genetic analysis of adrenal absence: agenesis and aplasia. Trends Endocrinol Metab, 16, 458–468.)
Overview of human adrenal development. A-C, The adrenogonadal primordium develops at around 4 weeks’ gestation, after which the adrenal primordium becomes a distinct structure that then migrates retroperitoneally to the cranial pole of the mesonephros. D, By 8 to 9 weeks’ gestation, the adrenal gland is encapsulated, contains chromaffin cells (black), and has distinct fetal (FZ) and definitive zones (DZ). Some of the signaling molecules, transcription factors and growth factors implicated in adrenal development are shown here, although the exact timing and interaction of many of these factors remains poorly understood at present. (Adapted with permission from Else, T., & Hammer, G. D. (2005). Genetic analysis of adrenal absence: agenesis and aplasia. Trends Endocrinol Metab, 16, 458–468.)
The fetal adrenal cortex consists of an outer “definitive” zone, the principal site of glucocorticoid and mineralocorticoid synthesis, and a much larger “fetal” zone that makes androgenic precursors (DHEA, DHEAS), which the placenta converts to estriol. A putative “transitional” zone exists between these regions toward the end of fetal development, but its role is unclear. The fetal adrenal glands are huge in proportion to other structures, and continue to grow well into the third trimester (Figure 13-2). At birth, the adrenals weigh 8 to 9 g, about the same size of adult adrenals, and represent approximately 0.4% of total body weight. However, the fetal adrenal zone rapidly involutes following birth and has virtually disappeared by 6 to 12 months of postnatal life. Thereafter, adrenal growth is comparatively slow so that the adrenal glands represent only 0.01% of body weight in the adult.
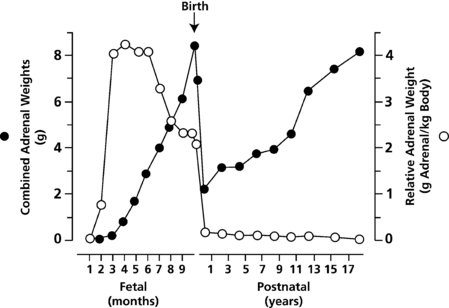
FIGURE 13-2  Combined adrenal weight (filled circles) and relative adrenal weight (open circles) from the first trimester through to early adulthood. (Reprinted with permission from Mesiano, S., & Jaffe, R. B. (1997). Developmental and functional biology of the primate fetal adrenal cortex. Endocr Rev, 18, 378–403.)
Combined adrenal weight (filled circles) and relative adrenal weight (open circles) from the first trimester through to early adulthood. (Reprinted with permission from Mesiano, S., & Jaffe, R. B. (1997). Developmental and functional biology of the primate fetal adrenal cortex. Endocr Rev, 18, 378–403.)
The complex mechanisms regulating adrenal development are still relatively poorly understood. However, important insight into key factors has been obtained from studies of transgenic mice and from patients with disorders of adrenal development.6 For example, the early stages of adrenal differentiation and development involve a number of signaling pathways (hedgehog/GLI3, WNT3/WNT4/WNT11, midkine), transcription factors (SALL1, FOXD2, PBX1, WT1, SF1 [NR5A1], DAX1 [NR0B1]), co-regulators (CITED2), matrix proteins (SPARC), and regulators of telomerase activity (ACD).7 Subsequent fetal adrenal growth is highly dependent on the tropic effects of adrenocorticotropin (ACTH), its receptor (MC2R), and its downstream signaling pathways, as well as growth factor signaling pathways such as insulin-like growth factor II (IGFII), basic fibroblast growth factor (bFGF), and epidermal growth factor (EGF).
Anatomy
The adrenals, once termed suprarenal glands, derive their name from their anatomic location, sitting on top of the upper pole of each kidney. Unlike most other organs, the arteries and veins serving the adrenal do not run in parallel. Arterial blood is provided by several small arteries arising from the renal and phrenic arteries, the aorta, and sometimes the ovarian and left spermatic arteries. The veins are more conventional, with the left adrenal vein draining into the left renal vein and the right adrenal vein draining directly into the vena cava. Arterial blood enters the sinusoidal circulation of the cortex and drains toward the medulla, so that medullary chromaffin cells are bathed in very high concentrations of steroid hormones. High concentrations of cortisol are required for the expression of medullary phenylethanolamine-N-methyltransferase, which converts norepinephrine to epinephrine, linking the adrenal cortical and medullary responses to stress.8
Steroid hormone synthesis
Early steps: Cholesterol uptake, storage, and transport
Much is now known about steroid biosynthesis,9 and the early steps in the intracellular trafficking of cholesterol have been reviewed.10 The human adrenal can synthesize cholesterol de novo from acetate, but most of its supply of cholesterol comes from plasma low-density lipoproteins (LDL) derived from dietary cholesterol. Rodent adrenals derive most of their cholesterol from high-density lipoproteins via a receptor termed SR-B1, but this pathway appears to play a minor role in human steroidogenesis. Adequate concentrations of LDL will suppress 3-hydroxy-3-methylglutaryl co-enzyme A (HMG CoA) reductase, the rate-limiting enzyme in cholesterol synthesis. ACTH, which stimulates adrenal steroidogenesis, also stimulates the activity of HMG CoA reductase, LDL receptors, and uptake of LDL cholesterol. LDL cholesterol esters are taken up by receptor-mediated endocytosis, then they are stored directly or converted to free cholesterol and used for steroid hormone synthesis. Cholesterol can be esterified by acyl-CoA:cholesterol transferase (ACAT), stored in lipid droplets, and accessed by activation of hormone-sensitive lipase (HSL) and by the so-called NPC proteins, which derive their name from their causative role in Niemann-Pick type C disease. ACTH stimulates HSL and inhibits ACAT, thus increasing the availability of free cholesterol for steroid hormone synthesis.
Steroidogenic enzymes
Cytochrome P450
Most steroidogenic enzymes are members of the cytochrome P450 group of oxidases.9 Cytochrome P450 is a generic term for a group of oxidative enzymes, all of which have about 500 amino acids and contain a single heme group. They are termed P450 (pigment 450) because all absorb light at 450 nm in their reduced states. It is sometimes stated that certain steroidogenic enzymes are “P450-dependent” enzymes. This is a misnomer, as it implies a generic P450 cofactor to a substrate-specific enzyme; however, the P450 binds the steroidal substrate and achieves its catalysis on an active site associated with the heme group. Human beings have genes for 57 cytochrome P450 enzymes, of which 7 are targeted to mitochondria and 50 are targeted to the endoplasmic reticulum, especially in the liver, where they metabolize countless endogenous and exogenous toxins, drugs, xenobiotics, and environmental pollutants. Each P450 enzyme can metabolize multiple substrates, catalyzing a broad array of oxidations. This theme recurs with each adrenal P450 enzyme.
Five distinct P450 enzymes are involved in adrenal steroidogenesis (Figure 13-3). Mitochondrial P450scc (CYP11A1) is the cholesterol side-chain cleavage enzyme catalyzing the series of reactions formerly termed 20,22 desmolase. Two distinct isozymes of P450c11, P450c11β (CYP11B1) and P450c11AS (CYP11B2), also found in mitochondria, catalyze 11β-hydroxylase, 18-hydroxylase, and 18-methyl oxidase activities. P450c17 (CYP17A1), found in the endoplasmic reticulum, catalyzes both 17α-hydroxylase and 17,20 lyase activities, and P450c21 (CYP21A2) catalyzes the 21-hydroxylation of both glucocorticoids and mineralocorticoids. In the gonads and elsewhere, P450aro (CYP19A1) in the endoplasmic reticulum catalyzes aromatization of androgens to estrogens.
FIGURE 13-3  Principal pathways of human adrenal steroid hormone synthesis. Other quantitatively and physiologically minor steroids are also produced. The names of the enzymes are shown by each reaction, and the traditional names of the enzymatic activities correspond to the circled numbers. Reaction 1: Mitochondrial cytochrome P450scc mediates 20α-hydroxylation, 22-hydroxylation, and scission of the C20-22 carbon bond. Reaction 2: 3βHSD mediates 3β-hydroxysteroid dehydrogenase and isomerase activities, converting Δ5 steroids to Δ4 steroids. Reaction 3: P450c17 catalyzes the 17α-hydroxylation of pregnenolone to 17OH-pregnenolone and of progesterone to 17OH-progesterone. Reaction 4: The 17,20 lyase activity of P450c17 converts 17OH-pregnenolone to DHEA; only insignificant amounts of 17OH-progesterone are converted to Δ4 androstenedione by human P450c17, although this reaction occurs in other species. Reaction 5: P450c21 catalyzes the 21-hydroxylation of progesterone to DOC and of 17OH-progesterone to 11-deoxycortisol. Reaction 6: DOC is converted to corticosterone by the 11-hydroxylase activity of P450c11AS in the zona glomerulosa and by P450c11β in the zona fasciculata. Reaction 7: 11-deoxycortisol undergoes 11β-hydroxylation by P450c11β to produce cortisol in the zona fasciculata. Reactions 8 and 9: The 18-hydroxylase and 18-methyl oxidase activities of P450c11AS convert corticosterone to 18OH-corticosterone and aldosterone, respectively, in the zona glomerulosa. Reactions 10 and 11 are found principally in the testes and ovaries. Reaction 10: 17βHSD-III converts DHEA to androstenediol and androstenedione to testosterone, whereas 17βHSD-I converts estrone to estradiol. Reaction 11: Testosterone may be converted to estradiol and androstenedione may be converted to estrone by P450aro.
Principal pathways of human adrenal steroid hormone synthesis. Other quantitatively and physiologically minor steroids are also produced. The names of the enzymes are shown by each reaction, and the traditional names of the enzymatic activities correspond to the circled numbers. Reaction 1: Mitochondrial cytochrome P450scc mediates 20α-hydroxylation, 22-hydroxylation, and scission of the C20-22 carbon bond. Reaction 2: 3βHSD mediates 3β-hydroxysteroid dehydrogenase and isomerase activities, converting Δ5 steroids to Δ4 steroids. Reaction 3: P450c17 catalyzes the 17α-hydroxylation of pregnenolone to 17OH-pregnenolone and of progesterone to 17OH-progesterone. Reaction 4: The 17,20 lyase activity of P450c17 converts 17OH-pregnenolone to DHEA; only insignificant amounts of 17OH-progesterone are converted to Δ4 androstenedione by human P450c17, although this reaction occurs in other species. Reaction 5: P450c21 catalyzes the 21-hydroxylation of progesterone to DOC and of 17OH-progesterone to 11-deoxycortisol. Reaction 6: DOC is converted to corticosterone by the 11-hydroxylase activity of P450c11AS in the zona glomerulosa and by P450c11β in the zona fasciculata. Reaction 7: 11-deoxycortisol undergoes 11β-hydroxylation by P450c11β to produce cortisol in the zona fasciculata. Reactions 8 and 9: The 18-hydroxylase and 18-methyl oxidase activities of P450c11AS convert corticosterone to 18OH-corticosterone and aldosterone, respectively, in the zona glomerulosa. Reactions 10 and 11 are found principally in the testes and ovaries. Reaction 10: 17βHSD-III converts DHEA to androstenediol and androstenedione to testosterone, whereas 17βHSD-I converts estrone to estradiol. Reaction 11: Testosterone may be converted to estradiol and androstenedione may be converted to estrone by P450aro.
Hydroxysteroid dehydrogenases
The hydroxysteroid dehydrogenases have molecular masses of about 35 to 45 kilodaltons, do not have heme groups, and require NAD+ or NADP+ as cofactors. Whereas most steroidogenic reactions catalyzed by P450 enzymes are due to the action of a single form of P450, each of the reactions catalyzed by hydroxysteroid dehydrogenases can be catalyzed by at least two, often very different, isozymes. Members of this family include the 3α- and 3β-hydroxysteroid dehydrogenases, the two 11β-hydroxysteroid dehydrogenases, and a series of 17β-hydroxysteroid dehydrogenases; the 5α-reductases are unrelated to this family. Based on their structures, these enzymes fall into two groups: the short-chain dehydrogenase-reductase (SDR) family, characterized by a “Rossman fold,” and the aldo-keto reductase (AKR) family, characterized by a triosephosphate isomerase (TIM) barrel motif.11 The SDR enzymes include 11β-HSDs 1 and 2, and 17β-HSDs 1, 2, 3, and 4; the AKR enzymes include 17β-HSD5, which is important in extra-glandular activation of androgenic precursors, and the 3α-hydroxysteroid dehydrogenases that participate in the so-called backdoor pathway of fetal androgen synthesis (discussed later). Based on their activities, it is physiologically more useful to classify them as dehydrogenases or reductases. The dehydrogenases use NAD+ as their cofactor to oxidize hydroxysteroids to ketosteroids, and the reductases mainly use NADPH to reduce ketosteroids to hydroxysteroids. Although these enzymes are typically bidirectional in vitro, they tend to function in only one direction in intact cells, with the direction determined by the cofactor(s) available.11
P450scc
Conversion of cholesterol to pregnenolone in mitochondria is the first, rate-limiting, and hormonally regulated step in the synthesis of all steroid hormones.9,10 This involves three distinct chemical reactions, 20α-hydroxylation, 22-hydroxylation, and scission of the cholesterol side-chain to yield pregnenolone and isocaproic acid. Because 20-hydroxycholesterol, 22-hydroxycholesterol, and 20, 22-hydroxycholesterol could all be isolated from bovine adrenals in significant quantities, it was previously thought that three separate enzymes were involved. However, a single protein, termed P450scc (where SCC refers to the side chain cleavage of cholesterol), encoded by a single gene (CYP11A1), on chromosome 15 catalyzes all the steps between cholesterol and pregnenolone. These three reactions occur on a single active site that is in contact with the hydrophobic bilayer membrane. Deletion of the gene for P450scc in the rabbit or mouse eliminates all steroidogenesis, indicating that all steroidogenesis is initiated by this one enzyme.
Transport of electrons to P450scc: Adrenodoxin reductase and adrenodoxin
P450scc functions as the terminal oxidase in a mitochondrial electron transport system.12 Electrons from NADPH are accepted by a flavoprotein, termed adrenodoxin reductase, that is loosely associated with the inner mitochondrial membrane. Adrenodoxin reductase transfers the electrons to an iron/sulfur protein termed adrenodoxin, which is found in the mitochondrial matrix or loosely adherent to the inner mitochondrial membrane. Adrenodoxin then transfers the electrons to P450scc (Figure 13-4). Adrenodoxin reductase and adrenodoxin serve as generic electron transfer proteins for all mitochondrial P450s, and not just for those involved in steroidogenesis; hence, these proteins are also termed ferredoxin reductase and ferredoxin. Adrenodoxin forms a 1:1 complex with adrenodoxin reductase, then dissociates, then subsequently reforms an analogous 1:1 complex with a mitochondrial P450 such as P450scc or P450c11, thus functioning as an indiscriminate diffusible electron shuttle mechanism. Adrenodoxin reductase is a membrane-bound mitochondrial flavoprotein that receives electrons from NADPH. The human genes for adrenodoxin reductase and adrenodoxin are expressed in all tissues, indicating they may have other roles. Human mutations in these genes have not been described.
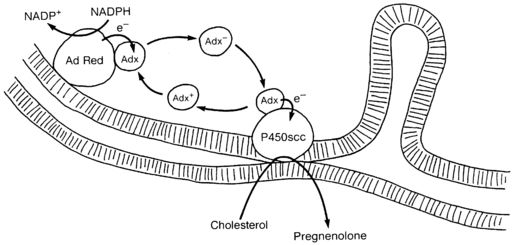
FIGURE 13-4  Electron transport to mitochondrial forms of cytochrome P450. Adrenodoxin reductase (AdRed), a flavoprotein loosely bound to the inner mitochondrial membrane, accepts electrons (e−) from NADPH, converting it to NADP+. These electrons are passed to adrenodoxin (Adx), an iron-sulfur protein in solution in the mitochondrial matrix that functions as a freely diffusible electron shuttle mechanism. Electrons from charged adrenodoxin (Adx−) are accepted by any available cytochrome P450, such as P450c11 or P450scc shown here. The uncharged adrenodoxin (Adx+) may then be again bound to adrenodoxin reductase to receive another pair of electrons. For P450scc, three pairs of electrons must be transported to the P450 to convert cholesterol to pregnenolone. The flow of cholesterol into the mitochondria is facilitated by StAR, which is not shown in this diagram. (Copyright W.L. Miller.)
Electron transport to mitochondrial forms of cytochrome P450. Adrenodoxin reductase (AdRed), a flavoprotein loosely bound to the inner mitochondrial membrane, accepts electrons (e−) from NADPH, converting it to NADP+. These electrons are passed to adrenodoxin (Adx), an iron-sulfur protein in solution in the mitochondrial matrix that functions as a freely diffusible electron shuttle mechanism. Electrons from charged adrenodoxin (Adx−) are accepted by any available cytochrome P450, such as P450c11 or P450scc shown here. The uncharged adrenodoxin (Adx+) may then be again bound to adrenodoxin reductase to receive another pair of electrons. For P450scc, three pairs of electrons must be transported to the P450 to convert cholesterol to pregnenolone. The flow of cholesterol into the mitochondria is facilitated by StAR, which is not shown in this diagram. (Copyright W.L. Miller.)
Mitochondrial cholesterol uptake: The steroidogenic acute regulatory protein, StAR
ACTH regulates steroidogenic capacity (chronic regulation) by inducing the transcription of genes for steroidogenic enzymes, but acute regulation, where steroids are released within minutes of a stimulus, is at the level of cholesterol access to P450scc.13,14 When either steroidogenic cells or intact rats are treated with inhibitors of protein synthesis such as cycloheximide, the acute steroidogenic response is eliminated, suggesting that a short-lived, cycloheximide-sensitive protein acts at the level of the mitochondrion as the specific trigger to the acute steroidogenic response. This factor was first identified as short-lived 30- and 37-kilodalton phosphoproteins that were rapidly synthesized when steroidogenic cells were stimulated with tropic hormones, then cloned from mouse Leydig MA-10 cells and named the steroidogenic acute regulatory protein, StAR.13,14 The central role of StAR in steroidogenesis was proven by finding that mutations of StAR caused congenital lipoid adrenal hyperplasia.15,16 Thus, StAR is the acute trigger that is required for the rapid flux of cholesterol from the outer to the inner mitochondrial membrane that is needed for the acute response of aldosterone to angiotensin II, of cortisol to ACTH, and of sex steroids to an LH pulse.
Some adrenal steroidogenesis is independent of StAR; when nonsteroidogenic cells are transfected with StAR and the P450scc system, they convert cholesterol to pregnenolone at about 14% of the StAR-induced rate.15,16 Furthermore, the placenta utilizes mitochondrial P450scc to initiate steroidogenesis but does not express StAR. The mechanism of StAR-independent steroidogenesis is unclear; it may occur without a triggering protein, or some other protein may exert StAR-like activity to promote cholesterol flux, but without StAR’s rapid kinetics. The exact mechanism of StAR’s action is unclear, but it is established that StAR acts on the outer mitochondrial membrane, does not need to enter the mitochondria to be active, and undergoes conformational changes on the outer mitochondrial membrane that are required for StAR’s activity.14,17 StAR functions as a component of a molecular machine termed a transduceosome on the outer mitochondrial membrane that consists of StAR, TSPO (the translocator protein formerly known as the peripheral benzodiazepine receptor), TSPO-associated protein 7 (PAP7, ACBD3 for acyl-CoA-binding-domain 3), the voltage-dependent anion channel (VDAC-1), and protein kinase A regulatory subunit 1α (PKAR1A).18 The precise fashion in which these proteins interact and move cholesterol from the OMM to P450scc and the means by which cholesterol is loaded into the OMM remain unclear. It is possible that new disorders of steroidogenesis will be described involving these proteins, but their mutation would be expected to affect many other systems as well.
3β-hydroxysteroid dehydrogenase/Δ5→Δ4 isomerase
Once pregnenolone is produced from cholesterol, it may undergo 17α-hydroxylation by P450c17 to yield 17-hydroxypregnenolone, or it may be converted to progesterone, the first biologically important steroid in the pathway. A single 42-kilodalton microsomal enzyme, 3β-hydroxysteroid dehydrogenase (3βHSD), catalyzes both conversion of the hydroxyl group to a keto group on carbon 3 and the isomerization of the double bond from the B ring (Δ5 steroids) to the A ring (Δ4 steroids). 9 Thus, a single enzyme, 3βHSD, converts pregnenolone to progesterone, 17α-hydroxypregnenolone to 17α-hydroxyprogesterone, dehydroepiandrosterone (DHEA) to androstenedione, and androstenediol to testosterone, all with the same enzymologic efficiency (Km and Vmax). As is typical of hydroxysteroid dehydrogenases, there are two isozymes of 3βHSD, encoded by separate genes (HSD3B1 and HSD3B2).19 These isozymes share 93.5% amino acid sequence identity and are enzymatically very similar. The enzyme catalyzing 3βHSD activity in the adrenals and gonads is the type 2 enzyme, whereas the type 1 enzyme, encoded by a closely linked gene with identical intron/exon organization, catalyzes 3βHSD activity in placenta, breast, and “extraglandular” tissues. Ultrastructural data surprisingly shows that bovine 3βHSD can be found in both the endoplasmic reticulum and in mitochondria. It is not clear if this is also true for human 3βHSD or if this subcellular distribution differs in various types of steroidogenic cells, but this could be a novel point regulating the direction of steroidogenesis.9
P450c17
Pregnenolone and progesterone may undergo 17α-hydroxylation to 17α-hydroxypregnenolone and 17α-hydroxyprogesterone (17OHP), respectively. 17OHP may also undergo scission of the C17,20 carbon bond to yield dehydroepiandrosterone (DHEA), but very little 17OHP is converted to androstenedione because the human P450c17 enzyme catalyzes this reaction at only 3% of the rate for conversion of 17α-hydroxypregnenolone to DHEA.20 These reactions are all mediated by a single enzyme, P450c17 (CYP17A1). This P450 is bound to smooth endoplasmic reticulum, where it accepts electrons from P450 oxidoreductase. As P450c17 has both 17α-hydroxylase activity and 17,20 lyase activity, it is the key branch point in steroid hormone synthesis. Neither activity of P450c17 is present in the adrenal zona glomerulosa, hence pregnenolone is converted to mineralocorticoids; in the zona fasciculata, the 17α-hydroxylase activity is present but 17,20 lyase activity is not, hence pregnenolone is converted to the glucocorticoid cortisol; in the zona reticularis, both activities are present, so that pregnenolone is converted to sex steroids (see Figure 13-3).9
17α-Hydroxylase and 17,20 lyase were once thought to be separate enzymes. The adrenals of prepubertal children synthesize ample cortisol but virtually no sex steroids (i.e., have 17α-hydroxylase activity but not 17,20 lyase activity), until adrenarche initiates the production of adrenal androgens (i.e., turns on 17,20 lyase activity). Furthermore, patients had been described lacking 17,20 lyase activity but retaining normal 17α-hydroxylase activity.21 However, studies of pig P450c17 showed that both 17α-hydroxylase and 17,20 lyase activities reside in a single protein, and cells transfected with a vector expressing P450c17 cDNA acquire both 17α-hydroxylase and 17,20 lyase activities. P450c17 is encoded by a single gene (CYP17A1) on chromosome 10q24.3 that is structurally related to the genes for P450c21 (21-hydroxylase).
Thus, the distinction between 17α-hydroxylase and 17,20 lyase is functional and not genetic or structural. Human P450c17 catalyzes 17α-hydroxylation of pregnenolone and progesterone equally well, but the 17,20 lyase activity of human P450c17 strongly prefers 17OH pregnenolone and not 17OH progesterone, consistent with the large amounts of dehydroepiandrosterone (DHEA) secreted by both the fetal and adult adrenal. Furthermore, the 17α-hydroxylase reaction occurs more readily than the 17,20 lyase reaction. The principal factor regulating the 17,20 lyase reaction is electron transport from NADPH.21
Electron transport to P450c17: P450 oxidoreductase and cytochrome b5
P450c17 (and P450c21) receive electrons from a membrane-bound flavoprotein, termed P450 oxidoreductase, which is a different protein from the mitochondrial flavoprotein, adrenodoxin reductase.12 P450 oxidoreductase receives two electrons from NADPH and transfers them one at a time to the P450. Electron transfer for the lyase reaction is promoted by the action of cytochrome b5 as an allosteric factor rather than as an alternate electron donor.20 17,20 Lyase activity also requires the phosphorylation of serine residues on P450c17 by a cAMP-dependent protein kinase21 (Figure 13-5). The availability of electrons determines whether P450c17 performs only 17α-hydroxylation or also performs 17,20 bond scission: increasing the ratio of P450 oxidoreductase or cytochrome b5 to P450c17 in vitro or in vivo increases the ratio of 17,20 lyase activity to 17α-hydroxylase activity. Competition between P450c17 and P450c21 for available 17-hydroxyprogesterone (17OHP) does not appear to be important in determining whether 17OHP undergoes 21-hydroxylation or 17,20 bond scission. Thus, the regulation of 17,20 lyase activity, and consequently of DHEA production, depends on factors that facilitate the flow of electrons to P450c17: high concentrations of P450 oxidoreductase, the presence of cytochrome b5, and serine phosphorylation of P450c17.21
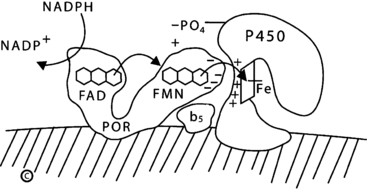
FIGURE 13-5  Electron transport to microsomal forms of cytochrome P450. NADPH interacts with P450 oxidoreductase (POR), bound to the endoplasmic reticulum, and gives up a pair of electrons (e−), which are received by the FAD moiety. Electron receipt elicits a conformational change, permitting the isoalloxazine rings of the FAD and FMN moieties to come close together, so that the electrons pass from the FAD to the FMN. Following another conformational change that returns the protein to its original orientation, the FMN domain of POR interacts with the redox-partner binding site of the P450. Electrons from the FMN domain of POR reach the heme group to mediate catalysis. The interaction of POR and the P450 is coordinated by negatively charged acidic residues on the surface of the FMN domain of POR and positively charged basic residues in the concave redox-partner binding site of the P450. The active site containing the steroid lies on the side of heme ring (Fe) opposite from the redox-partner binding site. In the case of human P450c17, this interaction is facilitated by the allosteric action of cytochrome b5 and by the serine phosphorylation of P450c17. (Copyright W.L. Miller.)
Electron transport to microsomal forms of cytochrome P450. NADPH interacts with P450 oxidoreductase (POR), bound to the endoplasmic reticulum, and gives up a pair of electrons (e−), which are received by the FAD moiety. Electron receipt elicits a conformational change, permitting the isoalloxazine rings of the FAD and FMN moieties to come close together, so that the electrons pass from the FAD to the FMN. Following another conformational change that returns the protein to its original orientation, the FMN domain of POR interacts with the redox-partner binding site of the P450. Electrons from the FMN domain of POR reach the heme group to mediate catalysis. The interaction of POR and the P450 is coordinated by negatively charged acidic residues on the surface of the FMN domain of POR and positively charged basic residues in the concave redox-partner binding site of the P450. The active site containing the steroid lies on the side of heme ring (Fe) opposite from the redox-partner binding site. In the case of human P450c17, this interaction is facilitated by the allosteric action of cytochrome b5 and by the serine phosphorylation of P450c17. (Copyright W.L. Miller.)
P450c21
After the synthesis of progesterone and 17-hydroxyprogesterone (17OHP), these steroids are hydroxylated at the 21 position to yield deoxycorticosterone (DOC) and 11-deoxycortisol, respectively9 (see Figure 13-3). The nature of the 21 hydroxylating step has been of great clinical interest because disordered 21-hydroxylation causes more than 90% of all cases of congenital adrenal hyperplasia. The clinical symptoms associated with this common genetic disease are complex and devastating.22 Decreased cortisol and aldosterone synthesis often lead to sodium loss, potassium retention, and hypotension, which will lead to cardiovascular collapse and death, usually within a month after birth if not treated appropriately. Decreased synthesis of cortisol in utero leads to overproduction of ACTH and consequent overstimulation of adrenal steroid synthesis; as the 21-hydroxylase step is impaired, 17OHP accumulates because P450c17 converts only miniscule amounts of 17OHP to androstenedione. However, 17-hydroxypregnenolone also accumulates and is converted to DHEA and subsequently to androstenedione and testosterone, resulting in severe prenatal virilization of female fetuses.22 Congenital adrenal hyperplasia (CAH) has been extensively studied clinically. Variations in the manifestations of the disease, especially the identification of patients without apparent defects in mineralocorticoid activity, suggested that there were two separate 21 hydroxylating enzymes that were differentially expressed in the zones of the adrenal specifically synthesizing aldosterone or cortisol. However, characterization of the P450c21 protein and gene cloning show there is only one 21-hydroxylase encoded by a single functional gene (CYP21A2) on chromosome 6p21.9,23,24 As this gene lies in the middle of the major histocompatibility locus, disorders of adrenal 21-hydroxylation are closely linked to specific HLA types.
Adrenal 21-hydroxylation is mediated by P450c21 found in smooth endoplasmic reticulum. P450c21 employs the same P450 oxidoreductase used by P450c17 to transport electrons from NADPH. 21-hydroxylase activity has also been described in a broad range of adult and fetal extra-adrenal tissues,25,26 especially in the liver, but is not catalyzed by P450c21.25 Hepatic 21-hydroxylation is mediated by several enzymes, notably CYP2C19 and CYP3A4, which are principally involved in drug metabolism; these enzymes can 21-hydroxylate progesterone, but not 17OHP, and hence may contribute to the synthesis of mineralocorticoids but not glucocorticoids.26,27
P450c11β and P450c11AS
Two closely related enzymes, P450c11β and P450c11as, catalyze the final steps in the synthesis of both glucocorticoids and mineralocorticoids.9,28,29 These two isozymes have 93% amino acid sequence identity and are encoded by tandemly duplicated genes (CYP11B1 and CYP11B2) on chromosome 8q21-22. Like P450scc, the two forms of P450c11 are found on the inner mitochondrial membrane and use adrenodoxin and adrenodoxin reductase to receive electrons from NADPH. By far the more abundant of the two isozymes is P450c11β, which is the classic 11β-hydroxylase that converts 11-deoxycortisol to cortisol and 11-deoxycorticosterone to corticosterone. The less abundant isozyme, P450c11AS, is found only in the zona glomerulosa, where it has 11β-hydroxylase, 18-hydroxylase, and 18-methyl oxidase (aldosterone synthase) activities; thus, P450c11AS is able to catalyze all the reactions needed to convert DOC to aldosterone.
P450c11β, which is principally involved in the synthesis of cortisol, is encoded by a gene (CYP11B1) that is primarily induced by ACTH via cAMP and is suppressed by glucocorticoids. The existence of two distinct functional genes is confirmed by the identification of mutations in each that cause distinct genetic disorders of steroidogenesis. Thus, patients with disorders in P450c11β have classic 11β-hydroxylase deficiency but can still produce aldosterone, whereas patients with disorders in P450c11AS have rare forms of aldosterone deficiency (so-called corticosterone methyl oxidase deficiency) while retaining the ability to produce cortisol.9,28,29
17β-hydroxysteroid dehydrogenase
Androstenedione is converted to testosterone, DHEA is converted to androstenediol, and estrone is converted to estradiol by the 17β-hydroxysteroid dehydrogenases (17βHSD; HSD17B), sometimes also termed 17-oxidoreductase or 17-ketosteroid reductase. The terminologies for these enzymes vary, depending on the direction of the reaction being considered.9,30–32 There is confusion in the literature about the 17βHSDs because (1) there are several different 17βHSDs; (2) some are preferential oxidases whereas others are preferential reductases; (3) they differ in their substrate preference and sites of expression; (4) there is inconsistent nomenclature, especially with the rodent enzymes; and (5) some proteins termed 17βHSD actually have very little 17βHSD activity and are principally involved in other reactions.
Type 1 17βHSD (17βHSD1), also known as estrogenic 17βHSD, is a 34-kilodalton cytosolic reductive SDR enzyme first isolated and cloned from the placenta, where it produces estriol, and is expressed in ovarian granulosa cells, where it produces estradiol.9,30–32 17βHSD1 uses NADPH as its cofactor to catalyze reductase activity. It acts as a dimer and only accepts steroid substrates with an aromatic A ring, so that its activity is confined to activating estrogens. The three-dimensional structure of human 17βHSD1 has been determined by x-ray crystallography. No genetic deficiency syndrome for 17βHSD1 has been described.
17βHSD2 is a microsomal oxidase that uses NAD+ to inactivate both estradiol to estrone and testosterone to Δ4 androstenedione. 17βHSD2 is found in the placenta, liver, small intestine, prostate, secretory endometrium, and ovary. In contrast to 17βHSD1, which is found in placental syncytiotrophoblast cells, 17βHSD2 is expressed in endothelial cells of placental intravillous vessels, consistent with its apparent role in defending the fetal circulation from transplacental passage of maternal estradiol or testosterone.9,30–32 No deficiency state for 17βHSD2 has been reported.
17βHSD3, the androgenic form of 17βHSD, is a microsomal enzyme that is apparently expressed only in the testis. This is the enzyme that is disordered in the classic syndrome of male pseudohermaphroditism that is often termed 17-ketosteroid reductase deficiency.9,30–32
An enzyme termed 17βHSD4 was initially identified as an NAD+-dependent oxidase with activities similar to 17βHSD2, but this peroxisomal protein is primarily an enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase.9 Deficiency of 17βHSD4 causes a form of Zellweger syndrome, in which bile acid biosynthesis is disturbed but steroidogenesis is not.
17βHSD5, originally cloned as a 3α-hydroxysteroid dehydrogenase, is an AKR enzyme (in contrast to 17βHSD types 1-4, which are SDR enzymes) termed AKR1C3 that catalyzes the reduction of Δ4 androstenedione to testosterone.9,33 The 17βHSD activity of 17βHSD5 is quite labile in vitro, and this enzyme catalyzes different activities under different conditions, which has led to confusion in its role in steroidogenesis.33 The adrenal zona reticularis expresses this enzyme at low levels, accounting for the small amount of testosterone produced by the adrenal.34
Steroid sulfotransferase and sulfatase
Steroid sulfates may be synthesized directly from cholesterol sulfate or may be formed by sulfation of steroids by cytosolic sulfotransferase (SULT) enzymes.35,36 At least 44 distinct isoforms of these enzymes have been identified, belonging to five families of SULT genes; many of these genes yield alternately spliced products accounting for the large number of enzymes. The SULT enzymes that sulfonate steroids include SULT1E (estrogens), SULT2A1 (nonaromatic steroids), and SULT2B1 (sterols). SULT2A1 is the principal sulfotransferase expressed in the adrenal, where it sulfates the 3β hydroxyl group of Δ5 steroids (pregnenolone, 17OH-pregnenolone, DHEA, androsterone) but not of cholesterol. SULT2B1a will also sulfonate pregnenolone but not cholesterol, whereas cholesterol is the principal substrate for SULT2B1b in the skin, liver, and elsewhere. It is not clear whether most steroid sulfates are simply inactivated forms of steroid or if they serve specific hormonal roles. Knockout of the mouse SULT1E1 gene is associated with elevated estrogen levels, increased expression of tissue factor in the placenta, and increased platelet activation, leading to placental thrombi and fetal loss that could be ameliorated by anticoagulant therapy. Mutations ablating the function of human SULT enzymes have not been described, but single nucleotide polymorphisms that alter the amino acid sequences and catalytic activity affecting drug activity are well described. African Americans have a high rate of polymorphisms in SULT2A1 apparently influencing plasma ratios of DHEA:DHEAS, which may correlate with risk of prostatic and other cancers.
To catalyze sulfation, SULT enzymes must receive sulfate in the form of 3’-phosphoadenosine-5’-phosphosulfate (PAPS). The production of PAPS requires PAPS synthase (PAPSS), which first converts ATP and sulfate (SO4) to adenosine phosphosulfate (APS), and then uses phosphate from another ATP molecule to convert APS to PAPS36. There are two human PAPSS genes: PAPSS1 is expressed ubiquitously, and PAPSS2 is abundantly expressed in adrenal and liver, where DHEA is sulfated36. PAPSS2 deficiency prevents DHEA sulfation so that the adrenal produces more non-sulfated DHEA than normal; this DHEA is converted to excess androgens. Compound heterozygous PAPSS2 deficiency was reported in a 6-year-old girl with premature pubarche and advanced bone age; by age 13 she had acne, hirsutism, and secondary amenorrhea37. Her serum concentrations of DHEA were high, DHEAS was very low, and androstendione, testosterone, and DHT were increased. She also had short stature and abnormal bone development, consistent with a role for PAPSS2 in cartilage and bone formation; complete PAPSS2 deficiency causes spondyloepimetaphyseal dysplasia.
Steroid sulfates may also be hydrolyzed to the native steroid-by-steroid sulfatase. Deletions in the steroid sulfatase gene on chromosome Xp22.3 cause X-linked ichthyosis. The fact that males have a single copy of this gene probably accounts for males having higher DHEAS levels than females of the same age. In the fetal adrenal and placenta, diminished or absent sulfatase deficiency reduces the pool of free DHEA available for placental conversion to estrogen, resulting in low concentrations of estriol in the maternal blood and urine. The accumulation of steroid sulfates in the stratum corneum of the skin causes the ichthyosis.
Aromatase: P450aro
Estrogens are produced by the aromatization of androgens, including adrenal androgens, by a complex series of reactions catalyzed by a single microsomal aromatase, P450aro.38,39 This typical cytochrome P450 is encoded by a single gene (CYP19A1) on chromosome 15q21.1. This gene uses several different promoter sequences, transcriptional start sites, and alternatively chosen first exons to encode aromatase mRNA in different tissues under different hormonal regulation. Aromatase expression in extraglandular tissues, especially adipose tissue, can convert adrenal androgens to estrogens. Aromatase in the epiphyses of growing bone converts testosterone to estradiol. The tall stature, delayed epiphyseal maturation, and osteopenia of males with aromatase deficiency, and their rapid reversal with estrogen replacement indicate that estrogen, not androgen, is responsible for epiphyseal maturation in males. Although it has traditionally been thought that aromatase activity is needed for embryonic and fetal development, infants and adults with genetic disorders in this enzyme have been described, showing that fetoplacental estrogen is not needed for normal fetal development.39
5α-reductase
Testosterone is converted to the more potent androgen, dihydrotestosterone, by 5α-reductase, an enzyme found in testosterone’s target tissues. There are two distinct forms of 5α-reductase. The type 1 enzyme, found in the scalp and other peripheral tissues, is encoded by a gene (SRD5A1) on chromosome 5; the type 2 enzyme, the predominant form found in male reproductive tissues, is encoded by a structurally related gene (SRD5A2) on chromosome 2p23.40 The syndrome of 5α-reductase deficiency, a disorder of male sexual differentiation, is due to a wide variety of mutations in the gene encoding the type 2 enzyme.41 The type 1 and 2 genes show an unusual pattern of developmental regulation of expression. The type 1 gene is not expressed in the fetus, then is expressed briefly in the skin of the newborn, and then remains unexpressed until its activity and protein are again found after puberty. The type 2 gene is expressed in fetal genital skin, in the normal prostate, and in prostatic hyperplasia and adenocarcinoma. Thus, the type 1 enzyme may be responsible for the pubertal virilization seen in patients with classic 5α-reductase deficiency, and the type 2 enzyme may be involved in male pattern baldness.40,41
11β-hydroxysteroid dehydrogenase
Although certain steroids are typically categorized as glucocorticoids or mineralocorticoids, the “mineralocorticoid” (glucocorticoid type 2) receptor has equal affinity for both aldosterone and cortisol. Nevertheless, cortisol does not act as a mineralocorticoid in vivo, even though cortisol concentrations can exceed aldosterone concentrations by 100- to 1000-fold, because mineralocorticoid responsive tissues (such as the kidney) convert cortisol to cortisone, a metabolically inactive steroid. The interconversion of cortisol and cortisone is mediated by two isozymes of 11β-hydroxysteroid dehydrogenase (11βHSD, HSD11B), each of which can catalyze both oxidase and reductase activity, depending on the cofactor available (NADP+ or NADPH).42–45 The ratio of NADP+ to NADPH is regulated by hexose-6-phosphate dehydrogenase (H6PDH).46 The type 1 enzyme (11βHSD1; HSD11B1) is expressed mainly in glucocorticoid-responsive tissues such as the liver, testis, lung, and proximal convoluted tubule. 11βHSD1 can catalyze both the oxidation of cortisol to cortisone using NADP+ as its cofactor (Km 1.6 μM), or the reduction of cortisone to cortisol using NADPH as its cofactor (Km 0.14 μM); the reaction catalyzed depends on which cofactor is available, but the enzyme can only function with high (micromolar) concentrations of steroid. 11βHSD2 (HSD11B2) catalyzes only the oxidation of cortisol to cortisone using NADH and can function with low (nanomolar) concentrations of steroid (Km 10 to 100 nM). 11βHSD2 is expressed in mineralocorticoid-responsive tissues and thus serves to “defend” the mineralocorticoid receptor by inactivating cortisol to cortisone, so that only “true” mineralocorticoids, such as aldosterone or deoxycorticosterone, can exert a mineralocorticoid effect. Thus, 11βHSD2 prevents cortisol from overwhelming renal mineralocorticoid receptors, and in the placenta and other fetal tissues 11βHSD2 also inactivates cortisol. The placenta also has abundant NADP+ favoring the oxidative action of 11βHSD1, so that in placenta both enzymes protect the fetus from high maternal concentrations of cortisol, but not from maternally administered betamethasone or dexamethasone. 11βHSD1 is located on the luminal side of the endoplasmic reticulum, and hence is not in contact with the cytoplasm. In this unusual cellular location, 11βHSD1 receives NADPH provided by H6PDH.44 This links 11βHSD1 to the pentose monophosphate shunt, providing a direct paracrine link between local glucocorticoid production and energy storage as fat.43–45
3α-hydroxysteroid dehydrogenases
The 3α-hydroxysteroid dehydrogenases (3αHSDs) are not familiar to most endocrinologists, but they are clinically important because of the discovery of the so-called backdoor pathway of steroidogenesis.47 This remarkable pathway (Figure 13-6), first discovered as the mechanism by which the male marsupial fetal testis makes androgens,47,48 plays a central role in human male sexual differentiation.49 In this pathway 17OHP is converted to dihydrotestosterone without going through DHEA, androstenedione, or testosterone, and hence it provides a mechanism for 17OHP to contribute to the virilization of female fetuses with 21-hydroxylase deficiency.22,50 There are four 3αHSD enzymes, sometimes termed types 1 through 4 but more properly termed AKR1C4-131; the numbering of the 3αHSD nomenclature is reversed in the AKR1C nomenclature, which is confusing and most unfortunate. These enzymes are structurally very similar, are encoded by a gene cluster on chromosome 10p14-15, and catalyze a wide array of steroidal conversions and other reactions.9 The backdoor pathway is characterized by both reductive and oxidative 3αHSD activities; the reductive activity can apparently be catalyzed by either AKR1C2 or AKR1C4.49 The nature of the oxidative activity remains uncertain, but may be catalyzed by retinol dehydrogenase (RoDH).9 AKR1C3, which converts androstenedione to testosterone in the adrenal, is also known as 17βHSD5. Further studies of the role of the backdoor pathway will be central to pediatric endocrinology.
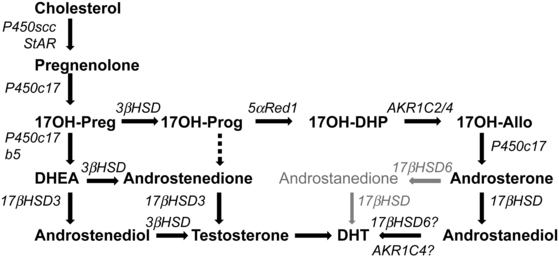
FIGURE 13-6  Pathways to androgens in CAH. In the absence of P450c21 activity, the adrenal can produce androgens by three pathways. First, the pathway from cholesterol to DHEA remains intact in 21-hydroxylase deficiency, and increased production of DHEA will lead to some DHEA being converted to androstenedione and thence to testosterone. Second, although minimal amounts of 17OHP are converted to androstenedione in the normal adrenal, the huge amounts of 17OHP produced in CAH permit some 17OHP to be converted to androstenedione and then to testosterone. Third, the so-called backdoor pathway depends on the 5α and 3α reduction of 17OHP to 17OH-allopregnenolone. This steroid is readily converted to androstanediol, which can then be oxidized to DHT by a 3αHSD enzyme, AKR1C2. Mass spectrometric examinations of human urinary steroids indicate this pathway is a major contributor in CAH.
Pathways to androgens in CAH. In the absence of P450c21 activity, the adrenal can produce androgens by three pathways. First, the pathway from cholesterol to DHEA remains intact in 21-hydroxylase deficiency, and increased production of DHEA will lead to some DHEA being converted to androstenedione and thence to testosterone. Second, although minimal amounts of 17OHP are converted to androstenedione in the normal adrenal, the huge amounts of 17OHP produced in CAH permit some 17OHP to be converted to androstenedione and then to testosterone. Third, the so-called backdoor pathway depends on the 5α and 3α reduction of 17OHP to 17OH-allopregnenolone. This steroid is readily converted to androstanediol, which can then be oxidized to DHT by a 3αHSD enzyme, AKR1C2. Mass spectrometric examinations of human urinary steroids indicate this pathway is a major contributor in CAH.
Fetal adrenal steroidogenesis
Adrenocortical steroidogenesis begins early in embryonic life, around 7 weeks after fertilization. Steroidogenic enzymes are immunocytochemically detected principally in the fetal zone at 50 to 52 days postconception, and by 8 weeks postconception the adrenal contains cortisol and responds to ACTH in primary culture systems.51 This cortisol synthesis is under the regulation of pituitary ACTH and involves transient expression of adrenal 3βHSD2; following the 9th week postconception, expression of 3βHSD2 and synthesis of cortisol wane; 3βHSD2 is barely detectable at 10 to 11 weeks and is absent at 14 weeks. At the same time, the fetal adrenal also produces 17βHSD5,51 which can convert androstenedione to testosterone. Thus, the fetal adrenal makes cortisol at the same time during gestation that fetal testicular testosterone is virilizing the genitalia of the normal male fetus. This fetal adrenal cortisol apparently suppresses ACTH, which otherwise would drive adrenal testosterone synthesis via 17βHSD5.
Fetuses affected with genetic lesions in adrenal steroidogenesis can produce sufficient adrenal androgen to virilize a female fetus to a nearly male appearance, and this masculinization of the genitalia is complete by the 12th week of gestation. The definitive zone of the fetal adrenal produces steroid hormones according to the pathways in Figure 13-3. By contrast, the large fetal zone of the adrenal is relatively deficient in 3βHSD2 activity after 12 weeks. The fetal adrenal has relatively abundant 17,20 lyase activity of P450c17; low 3βHSD and high 17,20 lyase activity account for the abundant production of dehydroepiandrosterone (DHEA) and its sulfate (DHEAS) by the fetal adrenal, which are converted to estrogens by the placenta (Figure 13-7). The fetal adrenal also has considerable sulfotransferase activity but little steroid sulfatase activity, also favoring conversion of DHEA to DHEAS. The resulting DHEAS cannot be a substrate for adrenal 3βHSD2; instead, it is secreted, 16α-hydroxylated in the fetal liver, and then acted on by placental 3βHSD1, 17βHSD1, and P450aro to produce estriol, or the substrates can bypass the liver to yield estrone and estradiol. Placental estrogens inhibit adrenal 3βHSD activity, providing a feedback system to promote production of DHEAS. Fetal adrenal steroids account for 50% of the estrone and estradiol and 90% of the estriol in the maternal circulation.
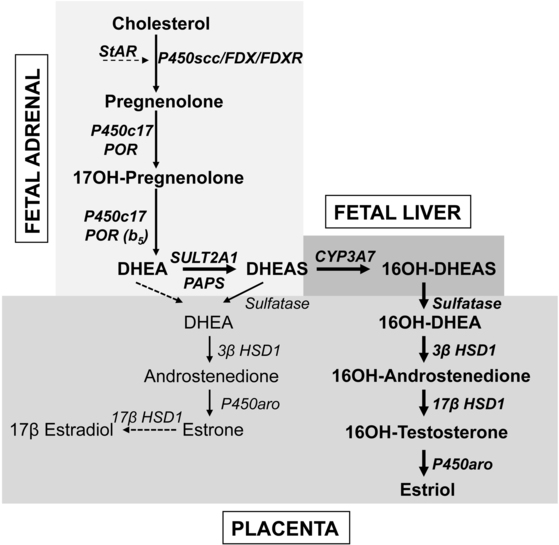
FIGURE 13-7  Steroid synthesis by the fetoplacental system. The fetal adrenal has minimal 3βHSD activity, hence the pathway from cholesterol to DHEA predominates. Most DHEA is converted to DHEAS by the sulfotransferase SULT2A1 and is then 16α-hydroxylated by CYP3A7 in the fetal liver. The 16α-hydroxy DHEAS reaches the placenta, where the sequential action of steroid sulfatase, 3βHSD1, 17βHSD1, and aromatase (P450aro) yield estriol, the principal steroidal product of the placenta. Small amounts of DHEA reach the placenta without being 16α-hydroxylated, where 3βHSD1 and P450aro can convert it to estrone; small amounts of estrone may also be converted to estradiol. About 80% of placental estrogen is estriol, 15% is estrone, and only 5% is estradiol.
Steroid synthesis by the fetoplacental system. The fetal adrenal has minimal 3βHSD activity, hence the pathway from cholesterol to DHEA predominates. Most DHEA is converted to DHEAS by the sulfotransferase SULT2A1 and is then 16α-hydroxylated by CYP3A7 in the fetal liver. The 16α-hydroxy DHEAS reaches the placenta, where the sequential action of steroid sulfatase, 3βHSD1, 17βHSD1, and aromatase (P450aro) yield estriol, the principal steroidal product of the placenta. Small amounts of DHEA reach the placenta without being 16α-hydroxylated, where 3βHSD1 and P450aro can convert it to estrone; small amounts of estrone may also be converted to estradiol. About 80% of placental estrogen is estriol, 15% is estrone, and only 5% is estradiol.
Although the fetoplacental unit produces huge amounts of DHEA, DHEAS, and estriol, as well as other steroids, they do not appear to serve an essential role.52 Successful pregnancy is wholly dependent on placental synthesis of progesterone, which suppresses uterine contractility and prevents spontaneous abortion; however, fetuses with genetic disorders of adrenal and gonadal steroidogenesis develop normally, reach term gestation, and undergo normal parturition and delivery. Mineralocorticoid production is only required postnatally, estrogens are not required, and androgens are only needed for male sexual differentiation. It appears that human fetal glucocorticoids are needed at about 8 to 12 weeks,51 but it is not clear that they are needed thereafter; if they are, the small amount of maternal cortisol that escapes placental inactivation suffices. A single newborn has been described with profound glucocorticoid resistance who was homozygous for a frameshift mutation at codon 772 in the glucocorticoid-binding domain of the glucocorticoid receptor.53 Although the infant had severe hypoglycemia and hypertension postnatally, pulmonary and other aspects of fetal development were normal, suggesting that glucocorticoid action is not required for normal human fetal development.
The regulation of steroidogenesis and growth of the fetal adrenal are not fully understood, but both are related to ACTH. ACTH effectively stimulates steroidogenesis by fetal adrenal cells in vitro, and excess ACTH is clearly involved in the adrenal growth and overproduction of androgens in fetuses affected with congenital adrenal hyperplasia. Experimental prenatal treatment of such fetuses by administering pharmacologic doses of dexamethasone to the mother at 6 to 10 weeks’ gestation can significantly reduce fetal adrenal androgen production and thus reduce the virilization of female fetuses, indicating that the hypothalamic-pituitary-adrenal (HPA) axis functions very early in fetal life.22 By contrast, anencephalic fetuses lacking pituitary ACTH have adrenals that contain a fairly normal complement of steroidogenic enzymes and retain their capacity for steroidogenesis. Thus, fetal adrenal steroidogenesis may be regulated by both ACTH-dependent and ACTH-independent mechanisms.
Regulation of steroidogenesis
The hypothalamic-pituitary-adrenal axis
Hypothalamus: CRF and AVP
The principal steroidal product of the human adrenal is cortisol, which is mainly secreted in response to adrenocorticotropic hormone (ACTH, corticotropin) produced in the pituitary; secretion of ACTH is stimulated primarily by corticotropin-releasing factor (CRF) from the hypothalamus. Hypothalamic CRF is a 41-amino-acid peptide synthesized mainly by neurons in the paraventricular nucleus. These same hypothalamic neurons also produce the decapeptide arginine vasopressin (AVP, also known as antidiuretic hormone or ADH).54 Both CRF and AVP are proteolytically derived from larger precursors with the AVP precursor containing the sequence for neurophysin, which is the AVP-binding protein. CRF and AVP travel through axons to the median eminence, which releases them into the pituitary portal circulation, although most AVP axons terminate in the posterior pituitary. AVP is co-secreted with CRF in response to stress, and both CRF and AVP stimulate the synthesis and release of ACTH, but they appear to do so by different mechanisms. CRF binds to a G-protein–coupled receptor on the membranes of pituitary corticotropes and activates adenylyl cyclase, increasing cAMP, which activates the protein kinase A (PKA) signaling pathway. PKA triggers ACTH secretion by concerted regulation of cellular potassium and calcium fluxes and enhances POMC gene transcription. AVP binds to its G-protein-coupled receptor and activates phospholipase C, which leads to the release of intracellular Ca++ and to the activation of protein kinase C (PKC). AVP seems to amplify the effects of CRF on ACTH secretion without affecting synthesis. However, CRF is the more important physiologic stimulator of ACTH release, although maximal doses of AVP can elicit a maximal ACTH response. When given together, CRF and AVP act synergistically, as would be expected from their independent mechanisms of action.
Pituitary: ACTH and POMC
Pituitary ACTH is a 39-amino-acid peptide derived from proopiomelanocortin (POMC), a 241-amino-acid protein.55 POMC undergoes a series of proteolytic cleavages, yielding several biologically active peptides (Figure 13-8). The N-terminal glycopeptide (POMC 1-75) can stimulate steroidogenesis and may function as an adrenal mitogen. POMC 112-150 is ACTH 1-39, POMC 112-126 and POMC 191-207 constitute α- and β-MSH (melanocyte stimulating hormone), respectively, and POMC 210-241 is β-endorphin. POMC is also produced in small amounts by the brain, testis, liver, kidney, and placenta, but this extrapituitary POMC does not contribute significantly to circulating ACTH. Malignant tumors will commonly produce “ectopic ACTH” in adults and rarely in children; this ACTH derives from ectopic biosynthesis of the same POMC precursor. Only the first 20 to 24 amino acids of ACTH are needed for its full biologic activity, and synthetic ACTH 1-24 is widely used in diagnostic tests of adrenal function. However, these shorter forms of ACTH have a shorter half-life than does native ACTH 1-39. POMC gene transcription is stimulated by CRF and inhibited by glucocorticoids.55
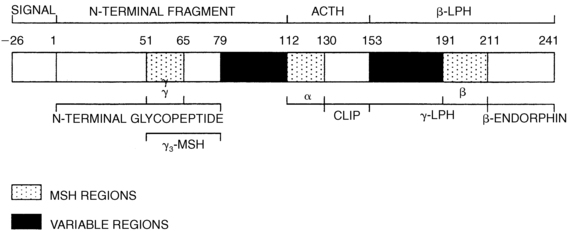
FIGURE 13-8  Structure of human prepro-opiomelanocortin. The numbers refer to amino acid positions, with No. 1 assigned to the first amino acid of POMC after the 26-amino-acid signal peptide. The α-, β-, and γ-MSH regions, which characterize the three “constant” regions, are indicated by stippling; the “variable” regions are solid. The amino acid numbers shown refer to the N terminal amino acid of each cleavage site; because these amino acids are removed, the numbers do not correspond exactly with the amino acid numbers of the peptides as used in the text. ACTH, adrenocorticotropic hormone; CLIP, corticotropin-like intermediate lobe peptide; LPH, lipotropic hormone; MSH, melanocyte-stimulating hormone.
Structure of human prepro-opiomelanocortin. The numbers refer to amino acid positions, with No. 1 assigned to the first amino acid of POMC after the 26-amino-acid signal peptide. The α-, β-, and γ-MSH regions, which characterize the three “constant” regions, are indicated by stippling; the “variable” regions are solid. The amino acid numbers shown refer to the N terminal amino acid of each cleavage site; because these amino acids are removed, the numbers do not correspond exactly with the amino acid numbers of the peptides as used in the text. ACTH, adrenocorticotropic hormone; CLIP, corticotropin-like intermediate lobe peptide; LPH, lipotropic hormone; MSH, melanocyte-stimulating hormone.
Actions of ACTH
ACTH stimulates the G-protein–coupled melanocortin 2 receptor (MC2R), which is located almost exclusively in the adrenal cortex. Activation of MC2R triggers the production of cAMP, activating PKA that catalyzes the phosphorylation of many proteins involved in steroidogenesis, thereby modifying their activity. ACTH elicits both acute and long-term effects. ACTH stimulates the biosynthesis of LDL receptors and the uptake of LDL, which provides most of the cholesterol used for steroidogenesis, and stimulates transcription of the gene for HMG-CoA reductase, the rate-limiting step in cholesterol biosynthesis, but adrenal biosynthesis of cholesterol is quantitatively much less important than the uptake of LDL cholesterol.10

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


