Lipid disorders in children and adolescents
Metabolism
Lipid disorders in children and adolescents can result from defects in the production, transport, or degradation of lipoproteins. To understand the diverse causes of lipoprotein abnormalities, a brief review of lipoprotein structure, function, and metabolism is provided. Table 23-1 summarizes the lipoprotein subclasses, the source of each one, and the constituent lipids and apolipoproteins associated with each particle.

Triglycerides, cholesterol esters, phospholipids, and plant sterols within food postingestion are digested to fatty acids, 2-monoglycerides, lysophospholipids, unesterified cholesterol, and plant sterols. Absorption of these digestive end products occurs through two mechanisms: passive diffusion and carrier-mediated transport. In passive diffusion, nonpolar lipids are solubilized with the aid of bile acids and lysophospholipids into mixed micelles that can diffuse through the apical surface of the enteric membrane. Carrier-mediated transport involves several different transport proteins for fatty acids and sterols. CD 36, a fatty acid translocase, promotes long chain fatty acid and cholesterol absorption in the proximal small intestine.1 At least two additional transporters, Niemann-Pick C1-like 1 protein (NPC1L1) and scavenger receptor B1 (SR-B1), play a role in sterol uptake.2,3 As such, NPC1L1 and SR-B1 are targets for the cholesterol-lowering medication ezetimibe, a potent inhibitor of cholesterol and plant sterol absorption.
Most of the plant sterol ingested and about half of the absorbed cholesterol are excreted from the intestinal cell back into the lumen by two adenosine triphosphate (ATP)-binding cassette (ABC) half-transporters, G5 and G8, thus limiting the amount of these sterols that is absorbed.4,5 A rare mutation of either ABCG5 or ABCG8, known as sitosterolemia, results in abnormally high plant sterol levels in plasma and tissues and deposition of sterols in the skin and arteries. Individuals with this disorder are at an increased risk of premature atherosclerosis.6
Within the enterocyte, lipids are aggregated into lipoproteins through the action of a microsomal triglyceride transfer protein (MTP). MTP conjugates triglycerides and cholesterol ester with apolipoprotein B-48 (apoB-48) on the luminal side of the endoplasmic reticulum (ER) membrane to create a mature chylomicron.7,8 A similar process is used to aggregate triglyceride and cholesterol with apoB-100 in the liver to form very low density lipoprotein (VLDL) particles. In the genetic disorder abetalipoproteinemia, mutations in the gene encoding MTP result in an inability to produce chylomicrons and VLDL, suggesting the essential nature of MTP in chylomicron and VLDL biogenesis.8
Chylomicrons once formed are too large to penetrate the capillary membrane. Consequently, they are secreted into the lymphatic system and enter the venous plasma compartment through the thoracic lymph duct. As the nascent particles are released into the plasma, several apolipoproteins (including apoC-II, C-III, and apoE) are preferentially transferred to the chylomicrons from circulating high-density lipoproteins (HDLs).9 Figure 23-1 depicts chylomicron metabolism.
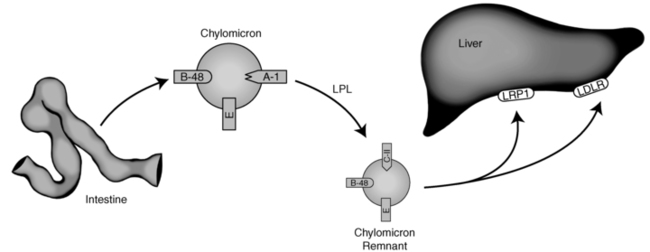
FIGURE 23-1  Endogenous lipoprotein metabolism. See text for details. (Courtesy of Emilie Graham, University of Cincinnati.)
Endogenous lipoprotein metabolism. See text for details. (Courtesy of Emilie Graham, University of Cincinnati.)
Chylomicrons transport dietary triglyceride and cholesterol to sites of storage or metabolism.10 They are rapidly cleared from the circulation through the action of lipoprotein lipase (LPL). LPL is a triglyceride hydrolase found on the capillary endothelium of various tissues, with its highest concentration in muscle and adipose tissues.11 LPL is activated by apoC-II and inhibited by apoC-III on the chylomicron. As the triglyceride contained within the chylomicron is hydrolyzed, the particle decreases in size. When approximately 80% of the initial triglyceride has been removed, apoC-II dissociates from its surface.11 The triglyceride-depleted chylomicrons, now considered chylomicron remnants, are taken up by the liver through the LDL receptor (LDLR), a receptor that recognizes apoE on the chylomicron surface and apoB100 on the surface of liver-derived lipoproteins.9 A smaller fraction of remnants may also be internalized via an LDLR-related protein-1 (LRP1)-mediated endocytosis.12,13
Very low density lipoproteins (VLDLs) originate from the liver, and like chylomicrons they are triglyceride-rich particles (Figure 23-2). In contrast to the intestinally derived chylomicrons, the fatty acids contained within the VLDL triglyceride come from de novo synthesis from dietary carbohydrate, lipoprotein remnants, or circulating fatty acids internalized by the liver from plasma.9 Within the hepatocyte, triglyceride and cholesterol ester are assembled by an MTP and surrounded with a phospholipid membrane associated with apoB-100.8 The mature VLDL particles are released into the lymph and ultimately into the vascular space, where other apolipoproteins (including apoC-II, apoC-III, and apoE) adsorb to the VLDL surface. The metabolism of the VLDL particle follows a route similar to that of the chylomicron: apoC-II on its surface activates LPL, LPL hydrolyzes the VLDL triglyceride, the particle decreases in size (after an 80% loss of triglyceride), and ultimately apoC-II dissociates—resulting in the formation of VLDL remnants (also known as intermediate-density lipoproteins [IDLs]). Approximately half of the IDL is then removed from plasma through the interaction of apoB with the LDLR on the surface of liver cells.9 The rest of the IDL is converted to LDL through further hydrolysis of core triglycerides by hepatic triglyceride lipase (HL).14 ApoE is transferred from IDL to HDL during the transition of the remnant to LDL.15
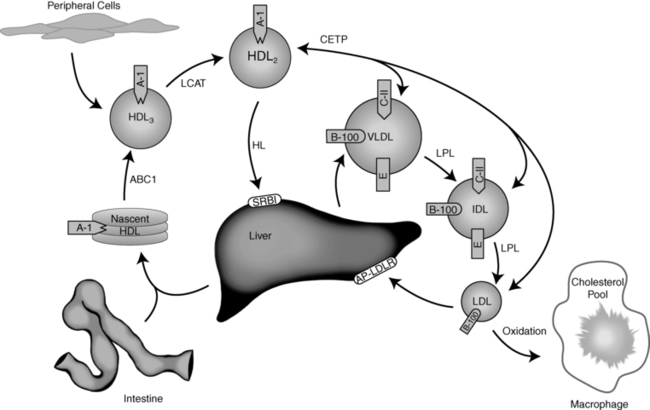
FIGURE 23-2  Exogenous lipoprotein metabolism. See text for details. (Courtesy of Emilie Graham, University of Cincinnati.)
Exogenous lipoprotein metabolism. See text for details. (Courtesy of Emilie Graham, University of Cincinnati.)
LDL, the major carrier of cholesterol in plasma, is taken up into peripheral tissues and liver cells by the LDLR assisted by an adaptor protein (AP). The AP binds to the LDLR and clathrin, suggesting a role for AP in the recruitment and retention of LDLR in clathrin-coated pits.16 Upon receptor binding, the LDL particle bound to LDLR/AP is rapidly internalized into clathrin-coated pits by endocytosis. Within the cell, the newly formed endosome becomes acidified through the action of an ATP-dependent proton pump.17 Acidification causes degradation of the clathrin coat, dissociation of the LDLR from LDL, and subdivision of the endosomal membranes. The endosome containing the LDLR recirculates back to the cell membrane for additional LDL uptake. Alternatively, proprotein convertase subtilisin/kexin type 9 (PCSK9), binds LDLR, and short-circuits recycling of LDLR from the endosome, leading to its degradation.18 The remaining LDL-containing endosome fuses with a lysosome, where hydrolytic enzymes digest the lipoprotein into its component parts: unesterified cholesterol, fatty acids, and free amino acids.17
The amount of cholesterol released from endosomal uptake regulates hepatic synthesis of LDLR and cholesterol. When cellular concentration of cholesterol is low, sterol receptor binding protein (SREBP) is released from the Golgi. SREBP translocates to the nucleus, where it serves as a nuclear factor that enhances the transcription of LDLR and hydroxymethylglutaryl (HMG) CoA reductase, the rate-limiting enzyme of cholesterol biosynthesis.19 In this way, intracellular hepatic cholesterol concentration regulates the amount of cholesterol internalized and synthesized by the cell.
When excess LDL and other small apoB-containing lipoproteins (chylomicron remnants and IDL) are present in the plasma, the capacity of the LDLR to remove them is exceeded and these particles become more susceptible to oxidation. Oxidized apoB-containing lipoproteins can be taken up by scavenger receptors on macrophages in the subendothelium of arteries and may contribute to the formation of atherosclerotic lesions.20
HDL transfers cholesterol and other lipids from peripheral tissues (including arterial atheroma) back to the liver. The particles are synthesized predominantly in the liver (and to a lesser extent in the intestine) as lipid-poor precursor particles (prebeta HDL) containing apoA-I (Figure 23-2).9 Nascent HDL interacts with the plasma membrane of cells, collecting lipid through an ATP-binding cassette transporter-A1 (ABCA1) mechanism.3,4 The cholesterol and phospholipids transferred through this process adsorb to the HDL, forming a disk-shaped particle referred to as HDL3. Within the plasma, HDL3 interacts with the enzyme lecithin cholesterol acyl transferase (LCAT)—which catalyzes the esterification of particle-associated cholesterol. ApoA-I on the HDL surface activates LCAT. Once formed, the cholesterol ester is more hydrophobic and moves to the interior of the particle—creating a sphere-shaped HDL particle known as HDL2.21
As HDL2 increases in size, the particle becomes substrate for cholesterol ester transfer protein (CETP). This enzyme promotes the exchange of esterified cholesterol within HDL2 for triglyceride contained within apoB-100–associated lipoproteins.22 This lipid exchange is the primary mechanism whereby HDL participates in reverse cholesterol transport from tissues back to the liver. The rest of the cholesterol ester is selectively taken up from HDL by hepatocytes via a SRB1, without concomitant uptake of the entire HDL particle. This latter process may require the action of HL.23 The lipid-poor prebeta HDL resulting from this process is released for recycling.16
Primary dyslipidemias
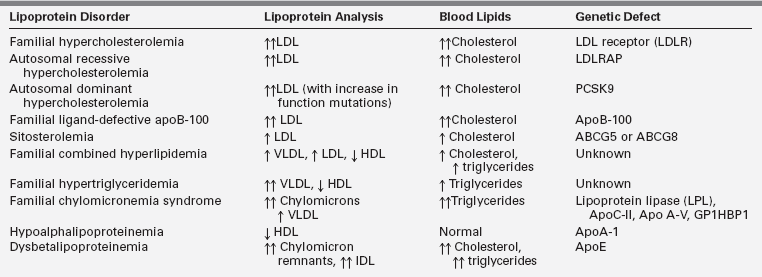
Lipoprotein synthesis, transport, and metabolism occur in many steps and involve many specialized proteins. A number of genetic defects have been identified in these processes and are referred to as primary dyslipidemias. Most of these genetic defects present in childhood. Table 23-2 summarizes pediatric lipoprotein disorders with reference to the characteristic lipoprotein profile of each one. The genetic and metabolic etiologies of these disorders are detailed in the following material.
Disorders of cholesterol metabolism
Familial hypercholesterolemia
Familial hypercholesterolemia (FH) is the most common single gene disorder of lipoprotein metabolism. FH is inherited as an autosomal-dominant trait with relatively low prevalence in Western countries. The prevalence has been reported to be 10 times higher in certain populations with a presumed founder effect, such as the Lebanese, the French Canadians, and the South Afrikaners.24 The heterozygous form is found in 1 in 500 persons, and the homozygous form is found in 1 in 1 million persons. The disorder is caused by a mutation in the LDLR gene.25 More than 1100 mutations in this gene have been identified, including those that affect receptor synthesis, intracellular transport, ligand binding, internalization, and recycling.26 In the heterozygous form, inheritance of one defective LDLR gene results in plasma LDL cholesterol levels two to three times higher than normal.25
Individuals with heterozygous FH are at an increased risk of developing early-onset coronary artery disease (CAD), usually between the ages of 30 and 60 years.24 In the homozygous form, individuals inherit a mutant allele for FH from both parents, resulting in plasma LDL cholesterol concentrations that are four to six times higher than normal.27 A more severe phenotype is found in individuals with receptor-negative mutations (those with <5% residual LDL receptor activity) compared to those with receptor-defective mutations (5% to 30% of normal LDL receptor activity).28 Due to the excessively high plasma cholesterol levels in individuals with homozygous FH, cholesterol deposits are common in the tendons (xanthomas) and eyelids (xanthelasmas)—generally by the age of 5 years.29 In the heterozygous form, xanthomas occur less frequently and generally not until one reaches older adulthood. Children with homozygous FH have early-onset atherosclerosis and often have myocardial infarction in the first decade of life, and death from CAD in the second decade. 29
Autosomal dominant and autosomal-recessive hypercholesterolemia
Autosomal-dominant hypercholesterolemia (ADH) is another inherited disorder resulting in a phenotype that is expressed as marked elevations or low levels of LDL cholesterol. ADH is caused by mutations in proprotein convertase subtilisin/kexin type 9 (PCSK9).30 This protein binds and favors degradation of the LDLR and thereby modulates the plasma levels of LDL cholesterol. Some of the naturally occurring PCSK9 mutations favor an increase in the function of the protein and cause hypercholesterolemia, whereas some favor a loss of function and are associated with low LDL cholesterol. The latter mutations appear to confer protection from developing CAD.
Autosomal recessive hypercholesterolemia (ARH) is caused by mutations in the ARH gene, which encodes the adaptor protein required for normal LDLR-mediated endocytosis in hepatocytes.31 Several different mutations in this protein have been identified, all leading to a lack of or suboptimal internalization of the LDLR.32 Cholesterol levels in individuals with ARH are five to six times higher than normal. Children with this disorder are clinically similar to those with homozygous FH. However, their parents usually have normal lipoprotein profiles. 31
Familial ligand-defective ApoB-100
Familial ligand-defective apoB-100 (FDB) is a monogenic disorder that clinically resembles heterozygous FH. The disease is characterized by moderate to markedly high plasma LDL cholesterol levels, normal triglycerides, and tendon xanthomas. The disorder is caused by poor binding of the LDL particle to the LDLR due to a mutation in apoB-100.33 Deficient LDLR binding results in a decreased clearance of LDL from plasma. The disorder is most common in individuals of European descent (1 per 1000).9 Patients with FDB are at moderate to high risk of developing CAD.34
Sitosterolemia
Sitosterolemia is a rare autosomal-recessive disease caused by a mutation in either of two genes (ABCG5 or ABCG8) encoding the ABC half-transporters.35 These genes are expressed in enterocytes and hepatocytes. The ABC half-transporters limit the absorption of cholesterol and plant sterols (and possibly shellfish sterols) in the gut. They also promote biliary and fecal excretion of cholesterol and phytosterols.36,37 Defective proteins result in an abnormally high absorption of plant sterols (and, to a lesser extent, cholesterol) into the enterocyte and decreased excretion of these sterols from the liver into the bile. Plasma cholesterol can be mildly, moderately, or markedly elevated, whereas plant sterol concentrations in the plasma are markedly increased. Patients with sitosterolemia develop premature CAD and xanthomas in childhood, and may develop aortic stenosis.35
Disorders of overproduction of VLDL
Familial combined hyperlipidemia (FCHL) is an autosomal-dominant disorder with a prevalence of 1% to 2% in Western populations.38 Individuals with FCHL generally share the same metabolic defect, which is overproduction of hepatic VLDL. Families with FCHL have multiple patterns of hyperlipidemia, including hypercholesterolemia, hypertriglyceridemia, and elevated apoB levels. A diagnosis of FCHL is based on the presence of increased levels of cholesterol, triglyceride, or apoB in patients and their first-degree relatives.38 Veerkamp and colleagues have developed a nomogram to calculate the probability that a person is likely to be affected by FCHL.39 A classic feature of FCHL is that the lipoprotein profile is variable in the same individual over time and may depend on factors such as diet, exercise, and weight. FCHL can manifest in childhood but is usually not fully expressed until adulthood.38 Patients with FCHL often have concurrent problems with insulin resistance, central obesity, and hypertension and are at an increased risk of premature CAD. 40
Syndromes with a similar phenotype are hyperapobetalipoproteinemia, LDL subclass pattern B, and the clustering of CVD risk factors known as metabolic syndrome in adults.38 Of the three, the latter syndrome is much more prevalent in children. Rates of metabolic syndrome are continuing to rise with the prevalence of obesity in the pediatric population.41 There appears to be a mechanistic link between central obesity, insulin resistance, and dyslipidemia—with central obesity generally preceding both glucose and lipid abnormalities.
Disorders of marked hypertriglyceridemia
Familial hypertriglyceridemia
Familial hypertriglyceridemia (FHTG) follows an autosomal-dominant inheritance pattern expressed predominantly in adulthood, with a population prevalence of ∼5% to 10%.38 The prevalence in children is increasing. Obesity is an important factor that can expedite the expression of FHTG, and patients often have concurrent glucose intolerance. The phenotype for FHTG is moderate to markedly high serum triglycerides (200 to 500 mg/dL range) and low to normal LDL and HDL cholesterol levels. The metabolic cause of the disorder is hepatic secretion of large triglyceride-rich VLDL particles that are catabolized slowly.42 The fundamental genetic defect for FHTG has not been identified.
Familial chylomicronemia syndrome
Chylomicronemia syndrome is a compilation of rare monogenetic disorders that cause marked impairment of lipoprotein lipase (LPL) activity. These disorders are phenotypically expressed as hypertriglyceridemia (usually triglycerides [TGs] > 1000 mg/dL) due to diminished or absent hydrolysis of chylomicron and VLDL-associated triglycerides by LPL.43 Impairment of LPL activity may be related to LPL deficiency, apoC-II (cofactor for LPL) deficiency, or the more recently described apoA5 and glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GPIHBP1) loss-of-function mutations that result in poor hydrolysis of chylomicron and VLDL-associated triglycerides.44
In homozygous chylomicronemia, fasting plasma has a viscous, creamy appearance due to the presence of large numbers of chylomicron particles. Risks for pancreatitis and hepatosplenomegaly are increased due to the markedly elevated serum triglycerides.43 In addition, eruptive xanthomas and neurologic symptoms may be apparent. Individuals heterozygous for the syndrome may have a mild to moderate elevation in plasma triglycerides. Environmental factors such as weight gain may exacerbate hypertriglyceridemia. Premature CAD is generally not a feature of chylomicronemia, but cases have been reported.44
Hypolipidemias
Low HDL cholesterol
In clinical practice, patients with low HDL cholesterol levels commonly have concurrent high triglycerides, with or without elevations in small dense LDL cholesterol.38 These patients are usually obese, and the mechanistic explanation for this dyslipidemic triad is VLDL overproduction. Less common are familial disorders of HDL, including familial hypoalphalipoproteinemia, mutations of the apoA-1 protein, Tangier disease, and LCAT deficiency.45 These disorders are characterized by a low HDL cholesterol level with no other lipid abnormality. Familial hypoalphalipoproteinemia follows an autosomal-dominant inheritance pattern.46 ApoA-1 levels are also often low due to decreased production of HDL.
A number of mutations have been described in the apoA-1 gene and are associated with low HDL cholesterol and low apoA-1.45,47 Tangier disease is due to mutations in the ABCA1 gene.47 Patients affected by this disease are not able to actively withdraw cholesterol from cells onto nascent HDL particles, causing rapid degradation of the nascent HDL. ApoA-1 is rapidly cleared before it is able to acquire cholesterol. In Tangier disease, HDL cholesterol levels are close to zero and the apoA-1 levels are less than 5 mg/dL. The risk of premature CAD in these patients is mild to moderate.46,47 LCAT deficiency is a very rare autosomal recessive disorder cause by mutations in LCAT, an enzyme synthesized by the liver and secreted into the plasma, where it associates with lipoproteins.48 LCAT esterifies free cholesterol on the surface of HDL and enables the accumulation of cholesteryl esters in the core of HDL. In LCAT deficiency, lack of normal cholesterol esterification impairs formation of mature HDL particles, which are readily catabolized along with apoA-1. Remarkably, despite the extremely low levels of plasma HDL cholesterol (usually < 10 mg/dL) and apoA-1, premature CAD is not a consistent feature of this disorder.
Abetalipoproteinemia
Abetalipoproteinemia is associated with low serum cholesterol (< 50 mg/dL) and triglycerides (∼2 to 45 mg/dL). Patients with this disorder present with steatorrhea and fatty liver. Without treatment, ataxia follows (with acanthocytosis and retinitis pigmentosa). Abetalipoproteinemia is caused by a defect in MTP.49 Without MTP, no chylomicrons, VLDL, or LDL appear in the plasma. In these patients, HDL takes over as the primary cholesterol carrier. Thus, the defect is not fatal. Because of significant fat malabsorption, fat-soluble vitamin status is impaired.
In particular, because vitamin E absorption and cellular uptake require chylomicron and LDL transport, high doses of vitamin E are required to prevent retinal and sensory neuron degeneration. Additional dietary considerations include restricting long-chain dietary triglycerides to less than 15 g/day to alleviate the steatorrhea. Medium-chain triglycerides (MCT oils) can be used as an alternative source of energy.44
Hypobetalipoproteinemia
Hypobetalipoproteinemia is an autosomal-dominant disorder resulting from a defect in the apoB gene that produces a truncated apolipoprotein B.50 Cholesterol levels in patients with heterozygous hypobetalipoproteinemia are usually 50% of those of an unaffected family member. The heterozygous form of this condition is benign. However, homozygous hypobetalipoproteinemia is associated with severe hypocholesterolemia, significant steatorrhea, fatty liver, acanthocytosis retinopathy, and peripheral neuropathy.51
Disorders with lipoprotein clearance via ApoE pathways
Dysbetalipoproteinemia is characterized by elevated cholesterol and triglyceride levels.38 The disorder results from the presence of a polymorphism of the apoE allele (apoE2, rather than the more common apoE3 or less common apoE4).52 Metabolically, this defect results in a poor uptake of remnant particles and abnormal remnant catabolism because of the abnormal apoE. Increased remnants, VLDL, chylomicrons, and apoE are all present. Xanthomas may occur, and premature CAD has been reported. This lipoprotein disorder is rare in children and often presents in young adulthood.38
Secondary causes
Secondary dyslipidemias can result from a variety of diseases and conditions (see Box 23-1). In the United States, the most prevalent cause of secondary dyslipidemia is overweight and obesity.53 The dyslipidemic triad (namely, elevated triglycerides and small dense LDL and low HDL cholesterol) is commonly associated with overweight (in particular, with central adiposity).53,54 In addition to dyslipidemia, insulin resistance and elevated blood pressure may be present. This cluster of abnormalities is known in adults as the metabolic syndrome. Empirical evidence in children also indicates that obesity during childhood is associated with the same risk factor clustering seen in adults, that it continues into adult life, and that it is associated with an increased risk for accelerated early atherosclerosis.55 The primary approach to treating this disorder in both adults and children is weight management. Improvement in weight status and a decrease in body fatness have been shown to be associated with improvements in the dyslipidemia and other comorbidities associated with obesity.56
Metabolic lipid perturbations in adult patients with types 1 and 2 diabetes mellitus are similar to those found in patients with the metabolic syndrome, but often are more severe.57 Generally in adults with diabetes, triglycerides are elevated and HDL cholesterol is low—and LDL cholesterol can be normal or mildly or moderately elevated. Diabetes in adults is considered a CAD risk equivalent according to the National Cholesterol Education Program (NCEP). This means that the risk for developing CAD in patients with poorly controlled diabetes is equivalent to those with established CAD.58 For this reason, the NCEP recommends aggressive treatment of dyslipidemia in adult patients with diabetes.
Although type 1 diabetes is currently the main form of diabetes seen in children, in the United States a growing number of patients with type 2 diabetes are under the age of 18 years.59 Change in the prevalence of type 2 diabetes in youth is likely related to the growing obesity epidemic occurring in the pediatric population.59,60 Data on lipid concentrations in children and adolescents with diabetes are few, particularly in those with type 2 diabetes.
The Search for Diabetes in Youth Study assessed the prevalence of serum lipid abnormalities among a representative sample of U.S. children and adolescents with type 1 and type 2 diabetes.61 Findings from this study showed a substantial number of diabetic children over the age of 10 years with abnormal serum lipids: nearly 50% had an LDL cholesterol level above the optimal level of 100 mg/dL. For children with type 2 diabetes, 37% had elevated triglyceride levels and 44% had low HDL cholesterol. These data highlight the importance of serum lipid screening in children with diabetes. A growing body of literature also shows early vascular dysfunction in children with diabetes, regardless of type.62 This is thought to be due to glycemic and lipid abnormalities associated with poorly managed diabetes. For this reason, new treatment guidelines recommend intensive glucose and lipid management for children with diabetes.56 These guidelines are discussed later in the chapter.
Other causes of secondary dyslipidemia include hypothyroidism, nephrotic syndrome, other renal diseases, liver diseases, and infection.63 The risk of development of atherosclerosis with these conditions is unknown but is likely proportionate to the length of exposure and extent of elevation in serum LDL cholesterol levels. Cardiovascular disease is common in patients with chronic renal insufficiency.64 Dyslipidemias can also result from the ingestion of a variety of medications. These medications include progestins, estrogens, androgens, anabolic steroids, corticosteroids, cyclosporine, and retinoids. Secondary causes of dyslipidemias should be identified by patient historical data and a careful physical examination.63 Laboratory tests (including thyroid, renal, and liver function panels) can confirm the diagnosis.
Vascular changes and dyslipidemia
It is well established that elevated concentrations of total cholesterol and LDL cholesterol in adult life are strong and reversible risk factors for CAD.58 Whether dyslipidemia during childhood contributes to atherosclerotic lesions in coronary and other arteries has been a subject of debate, but accumulating evidence from pathology and in vivo imaging studies favors a relationship. Atherosclerotic lesions result from deposits of lipid and cholesterol in the intima of the arterial wall.65 Early lesions, called fatty streaks, are formed from the accumulation of macrophages filled with lipid droplets (foam cells).
Fatty streaks do not disorganize the normal structure of the intima, do not deform or obstruct the artery, and are in and of themselves not considered harmful.66 However, some continue to accumulate macrophage foam cells and extracellular lipid and smooth muscle cells—forming raised plaques. From these, more advanced lesions may develop—with further deposition of extracellular lipid, cholesterol crystals, collagen, and potentially calcium.67 It is these raised lesions that result in a myocardial infarction because of their increasing size and obstruction of the arterial lumen or because of rupture of the fibrous plaque, which results in the release of thrombogenic substances from the necrotic core.67
Pathobiologic studies of the coronary arteries of young individuals who died from causes unrelated to heart disease have been useful in documenting the progression of atherosclerosis by age and risk factor determinants. Stary and colleagues studied more than 500 postmortem samples of coronary arteries from persons younger than 30 years of age and found the presence of fatty streaks in the majority of children younger than 9 years of age, raised lesions in about half of adolescents, and more advanced lesions in about a third of the young adults studied.68 In 93 autopsies of young adults for whom childhood risk factor data were available, Berenson and colleagues found that the extent of the surface of arteries covered with fatty streaks and fibrous plaques was positively associated with LDL cholesterol, triglycerides, blood pressure, and body mass index and negatively associated with HDL cholesterol levels in childhood.69
The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study reached similar conclusions from examination of more than 3000 postmortem samples of coronary arteries of young adults who died from noncardiovascular events and who likewise had a variety of surrogates for antimortem risk factor measures available.70 In general, pathology studies have made important contributions to the identification of risk factors for early aspects of the atherosclerotic process. In conjunction with findings from longitudinal studies such as the Framingham Heart Study (in which risk factor assessments of participants preceded the development of cardiovascular disease),71 a group of risk factors, often referred to as the traditional risk factors for CAD, has been established. A complete list of pediatric risk factors for CAD is found in Box 23-2.
BOX 23-2 Pediatric Risk Factors for Coronary Artery Disease




