Tumor suppressor genes
David Cosgrove, MB, BCh  Ben Ho Park, MD, PhD
Ben Ho Park, MD, PhD  Bert Vogelstein, MD
Bert Vogelstein, MD
Overview
Cancer is a genetic disease. Mutations and other alterations in growth promoting genes (oncogenes) and tumor suppressor genes can accumulate during the lifetime of a normal cell resulting in cancer. Unlike oncogenes, tumor suppressor genes generally require biallelic inactivation in order to demonstrate a cancerous phenotype. Importantly, the discovery of heritable tumor suppressor gene mutations that lead to familial forms of cancer has revealed great insight into tumor suppressor function. This has clinical screening implications for individuals with a family history of cancer and has led to newer therapies to target cancer cells with loss of specific tumor suppressors.
A genetic basis for the development of cancer has been hypothesized for over a century, supported by familial, epidemiologic, and cytogenetic studies. However, only in the past 40 years has definitive evidence emerged that cancer is a genetic disease. It is now known that cancers arise through a multistage process in which inherited and somatic mutations of cellular genes lead to repeated waves of clonal selection in which cells with the most robust and aggressive growth properties prevail. Two classes of genes, oncogenes and tumor suppressor genes, are primary targets for the mutations as these genes control the ratio of cell birth and cell death. In all normal tissues of the adult, this ratio is exactly 1.0; mutations increase the ratio. A third class of genes, called genomic stability genes, do not alter the ratio when mutated, but it can indirectly contribute to tumorigenesis through an acceleration of mutations in oncogenes and tumor suppressor genes.
The vast majority of the mutations that contribute to the development and behavior of cancer cells are somatic (i.e., arising during tumor development) and are present only in the neoplastic cells of the individual. A small fraction of mutations in cancer cells are constitutional, present in all somatic cells and can be passed on to progeny, increasing cancer risk in subsequent generations.
The identification and function of oncogenes are reviewed in other chapters of this book. We provide a brief summary of their general properties as a comparison to tumor suppressor genes. In general, oncogenes have critical roles in a variety of growth regulatory pathways, and their protein products are distributed throughout many subcellular compartments. The mutant alleles present in cancers have sustained gain-of-function alterations resulting from point mutations, chromosomal rearrangements, gene amplifications, or other changes to the oncogene sequence. In the overwhelming majority of cancers, mutations in oncogenes arise somatically in the tumor cells, although germline mutations have been described, notably in RET (rearranged during transfection) and MET (metastasis), leading to familial medullary thyroid cancer and hereditary papillary renal cell carcinoma, respectively.
Although oncogenic alleles harbor activating mutations, tumor suppressor genes are defined by their inactivation in human cancer, and, as with oncogenes, the cellular functions of tumor suppressor genes appear to be diverse.
Defects in genomic stability genes have also been implicated in a broad spectrum of human cancers. Like tumor suppressor genes, the genomic stability genes are inactivated in human cancers. However, unlike the mutations in tumor suppressor genes, mutations in genomic stability genes are more often inherited in mutant form. For example, inherited mutations in the breast cancer 1 (BRCA1) early-onset or breast cancer 2 (BRCA2) early-onset genes play a key role in hereditary breast and ovarian cancers, but these genes are rarely mutated somatically in nonfamilial forms of breast cancers.
Enormous progress has been made in the identification of inherited and somatic mutations in oncogenes, tumor suppressor genes, and genomic stability genes in human cancers. The function of these genes has been elucidated, in part, through the analysis of a variety of model systems employing mice, flies, worms, and other organisms, and through the investigation of human cancer cell lines and sequencing of human cancer cells. The principal aims of this chapter are to review the somatic cell genetic and epidemiologic studies that established the existence of tumor suppressor genes; to describe the identification and cloning of representative tumor suppressor genes; to highlight selected studies of the function of tumor suppressor genes in the regulation of cell birth and cell death; and to illustrate an example of a genomic stability gene that plays a causal role in common human cancers.
Genetic basis for tumor development
The inherited basis of human cancer has been appreciated for almost 150 years. In 1866, Broca described a family in which many members developed breast or liver cancer, and he proposed that an inherited abnormality within the affected tissue allowed for tumor development.1 Following the rediscovery of Mendel’s work, studies of the rates of spontaneous mammary tumor formation in various inbred strains of mice led Haaland to argue that tumorigenesis could behave in a formal sense as a Mendelian genetic trait.2 Similarly, Warthin’s analysis of the pedigrees of cancer patients at the University of Michigan Hospital between 1895 and 1913 identified four multigenerational families with susceptibilities to specific cancer types that appeared to be transmitted as autosomal dominant Mendelian traits (Figure 1).3 Although these and other studies suggested the existence of an inherited genetic basis for some cancers, other explanations for familial clustering were certainly possible (e.g., shared exposure to a carcinogenic agent in the environment or diet). Furthermore, it was highlighted that most cancers in humans appeared to arise as sporadic, isolated cases.
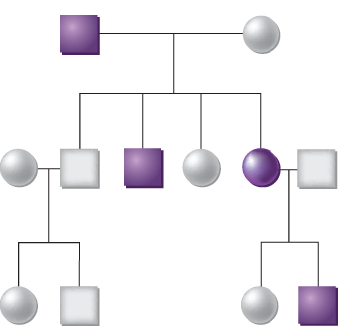
Figure 1 The inheritance of cancer in a family. The affected members with cancer are indicated by shaded squares (males) or circles (females). This family demonstrates a dominant pattern of inheritance, meaning that each offspring has a 50% chance of inheriting a germline mutation that predisposes to a high probability of developing cancer.
A role for somatic mutations in the development of cancer was first proposed by Boveri,4 who noted that, in sea urchin eggs fertilized by two sperms, abnormal mitotic divisions leading to the loss of chromosomes occurred in daughter cells, and atypical tissue masses could be seen in the resulting gastrula. He believed that these abnormal tissues appeared physically similar to the poorly differentiated tissue masses seen in tumors and hypothesized that cancer arose from a cellular aberration producing abnormal mitotic figures. Boveri’s hypothesis apparently did not gain favor at the time, initially because of the lack of direct experimental support from studies of the karyotypes of animal and human tumors. Such karyotypes were impossible to obtain with the available technology. Once the karyotypic studies were performed decades later, and appeared to support his hypothesis, Boveri was still doubted because of uncertainty about whether the changes in chromosome number in tumors were a cause or an effect of the neoplastic process.
A landmark observation in the search to identify a genetic basis for cancer was reported by Rous in 1911, when he showed that sarcomas could be reproducibly induced in chickens by cell-free filtrates of a sarcoma that had previously arisen in another chicken.5 Although this observation provided strong evidence that neoplasms could be virally induced, the observation also provided support for the view that cancer could be attributed to discrete genetic elements. Sixty years after Rous’s initial report, the oncogenic region of the Rous sarcoma virus was identified. Further characterization and cloning of the transforming sequences demonstrated that the oncogenicity of the virus depended on vsrc, a transduced and mutated copy of the csrc cellular oncogene. Subsequently, all oncogenes of acutely transforming ribonucleic acid (RNA) tumor viruses have been found to be transduced cellular genes. (In fact, they were defined as oncogenes.) The viral oncogenes cause transformation because they are mutated versions of cellular oncogenes or are expressed at abnormally high levels. In human cancers, somatic mutations generate oncogenic alleles from oncogenes.
Oncogenes play a role in most forms of human cancer, but are particularly prominent in “liquid” tumors, such as leukemias and lymphomas, as well as in sarcomas. Such cancers often have characteristic chromosomal translocations that alter oncogenes at the breakpoints, fusing them with unrelated genes and endowing the fusion product with new properties that increase cell birth or decrease cell death.
Somatic cell genetic studies of tumorigenesis
The studies of Ephrussi et al.6 and Harris7 provided compelling evidence that the ability of cells to form a tumor behaves as a recessive trait at the cellular level. They observed that the growth of murine tumor cells in syngeneic animals could be suppressed when the malignant cells were fused to nonmalignant cells, although reversion to tumorigenicity often occurred when the hybrids were propagated for extended periods in culture. The reappearance of malignancy was found to be associated with specific chromosome losses. The interpretation of those authors, that malignancy can be suppressed in somatic cell hybrids, was subsequently supported by additional studies on mouse, rat, and hamster intraspecies somatic cell hybrids, as well as by interspecies hybrids between rodent tumor cells and normal human cells.8, 9 The karyotypic instability of the rodent human hybrids, however, complicated the analysis of the human chromosomes involved in the suppression process. Stanbridge and his colleagues overcame this problem by studying hybrids made by fusing human tumor cell lines to normal, diploid human fibroblasts.10, 11 Their analysis confirmed that hybrids retaining both sets of parental chromosomes were suppressed, with tumorigenic variants arising only rarely after chromosome losses in the hybrids. Moreover, it was demonstrated that the loss of specific chromosomes, and not simply chromosome loss in general, correlated with the reversion to tumorigenicity. Tumorigenicity could be suppressed even if activated oncogenes, such as mutant ras genes, were expressed in the hybrids.11, 12
The observation that the loss of specific chromosomes was associated with the reversion to malignancy suggested that a single chromosome (and perhaps even a single gene) might be sufficient to suppress tumorigenicity. To directly test that hypothesis, the technique of microcell-mediated chromosome transfer was used to transfer single chromosomes from normal cells to tumor cells. It was found that the transfer of a single chromosome 11 into the HeLa cervical carcinoma cell line suppressed the tumorigenic phenotype of the cells.13 Similarly, transfer of chromosome 11 into a Wilms tumor cell line was found to suppress tumorigenicity, whereas the transfer of several other chromosomes had no effect.14 Many studies have demonstrated that transfer of even very small chromosome fragments will specifically suppress the tumorigenic properties of certain cancer cell lines.
Although tumorigenic growth in immunocompromised animals can often be suppressed in hybrids resulting from fusion between malignant and normal cells or by transfer of unique chromosome fragments, other traits characteristic of the parental tumor cells, such as immortality and anchorage-independent growth in vitro, may be retained. This observation is consistent with the notion that most malignant tumors arise as a result of multiple genetic alterations. Suppression of tumorigenicity following cell fusion or microcell chromosome transfer might thus represent correction of only one of many alterations.
In summary, somatic cell genetic approaches provided early and persuasive evidence for the existence of critical growth-regulating genes in normal cells that can suppress phenotypic traits of immortal or even fully cancerous cells.
Retinoblastoma: a paradigm for tumor suppressor gene function
Essentially concurrent with the initial cell fusion experiments of Harris and colleagues, Knudson’s analysis of the age-specific incidence of retinoblastoma led him to propose that two “hits” or mutagenic events were necessary for retinoblastoma development.15 Retinoblastoma occurs sporadically in most cases, but, in some families, it displays an autosomal dominant inheritance. In an individual with the inherited form of the disease, Knudson proposed that the first hit is present in the germline, and thus in all cells of the body. However, the presence of a mutation at the susceptibility locus was argued to be insufficient for tumor formation, and a second somatic mutation was hypothesized to be necessary for promoting tumor formation. Given the high likelihood of a somatic mutation occurring in at least one retinal cell during development, the dominant inheritance pattern of retinoblastoma in some families could be explained. In the nonhereditary form of retinoblastoma, both mutations were proposed to arise somatically within the same cell. Although each of the two hits could theoretically have been in different genes, subsequent studies (see later) led to the conclusion that both hits were at the same genetic locus, ultimately inactivating both alleles of the retinoblastoma 1 (RB1) susceptibility gene. Knudson’s hypothesis served not only to illustrate mechanisms through which inherited and somatic genetic changes might collaborate in tumorigenesis but also to link the notion of recessive genetic determinants for human cancer to somatic cell genetic findings on the recessive nature of tumorigenesis.
The first clue to the location of a putative gene responsible for inherited retinoblastoma was obtained from karyotypic analyses of patients with retinoblastoma. Constitutional deletions of chromosome 13 were observed in some cases.16 Subsequent cytogenetic studies of patients with retinoblastoma identified detectable germline deletions of chromosome 13 in only about 5% of all patients. However, in cases where deletions were observed, the common region of deletion was centered around chromosome band 13q14.17 When compared with karyotypically normal family members, patients with deletions of 13q14 were found to have reduced levels of esterase D, an enzyme of unknown physiologic function.18 This finding implied that the esterase D gene might be contained within chromosome band 13q14. Indeed, analysis of the segregation patterns of esterase D isozymes and retinoblastoma development in families with inherited retinoblastoma established that the esterase D and RB1 loci were genetically linked.19
Subsequently, a child with inherited retinoblastoma was found to have esterase D levels approximately one-half of normal, although no deletion of chromosome 13 was seen in karyotype studies of his blood cells and skin fibroblasts.20 Interestingly, tumor cells from this patient had a complete absence of esterase D activity, despite harboring one apparently intact copy of chromosome 13. Based on these findings, it was proposed that the copy of chromosome 13 retained in the tumor cells had a submicroscopic deletion of both the esterase D and RB1 loci. Moreover, it was concluded that the initial RB1 mutation in the child was recessive at the cellular level (i.e., cells with inactivation of one RB1 allele had a normal phenotype). However, the effect of the predisposing mutation could be unmasked in the tumor cells by a second event, such as the loss of the chromosome 13 carrying the wild-type RB1 allele. This proposal was entirely consistent with Knudson’s two-hit hypothesis.15, 21
To establish the generality of these observations, Cavenee and colleagues undertook studies of retinoblastomas, both inherited and sporadic, by using deoxyribonucleic acid (DNA) probes from chromosome 13. Probes detecting DNA polymorphisms were used, so that the two parental copies of chromosome 13 in the cells of the patient’s normal and tumor tissues could be distinguished from one another. By using such markers to compare paired normal and tumor samples from each patient, the Cavenee group was able to demonstrate that loss of heterozygosity (LOH—i.e., the loss of one parental set of markers) for chromosome 13 alleles had occurred during tumorigenesis in more than 60% of the cases studied.22 LOH for chromosome 13, and specifically for the region of chromosome 13 containing the RB1 gene, occurred via a number of different mechanisms (Figure 2). In addition, through study of inherited cases, it was shown that the copy of chromosome 13 retained in the tumor cells was derived from the affected parent and that the chromosome carrying the wild-type RB1 allele had been lost.22, 23 These data established that the unmasking of a predisposing mutation at the RB1 gene, whether the initial mutation had been inherited or had arisen somatically in a single developing retinoblast, occurred by the same chromosomal mechanisms.
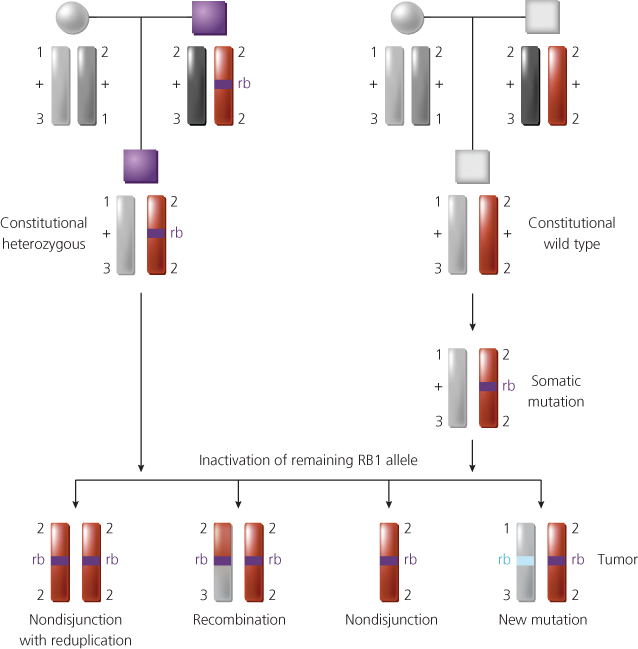
Figure 2 Chromosomal mechanisms that result in loss of heterozygosity for alleles at the retinoblastoma predisposition (RB1) locus at chromosomal band 13q14. In the inherited form of the disease (top left), the affected son inherits a mutant RB1 allele (rb) from his affected father and a normal RB1 allele (+) from his mother. Thus, he has one wild-type and one mutant RB1 allele in all his cells (i.e., constitutional genotype for RB1 is rb/+). The two copies of chromosome 13 in his normal cells (one from each parent) can be distinguished using polymorphic DNA markers flanking the RB1 locus (the polymorphic alleles are designated by number). A retinoblastoma can arise after inactivation of the remaining wild-type RB1 allele. Among the genetic mechanisms found to inactivate the remaining wild-type RB1 allele during tumor development are chromosome nondisjunction and reduplication of the remaining copy of chromosome 13, mitotic recombination, nondisjunction, and new RB mutations that inactivate the remaining RB1 allele. Shown at the top right is the situation in the noninherited (sporadic) form of the disease. A somatic mutation arises in a developing retinal cell and inactivates one of the RB1 alleles. A retinoblastoma will develop if the remaining RB1 allele is inactivated by one of the mechanisms shown.
Patients with the inherited form of retinoblastoma were known to be at an increased risk for the development of a few other cancer types, particularly osteosarcomas. LOH for the chromosome 13q region containing the RB1 locus was seen in osteosarcomas arising in patients with the inherited form of retinoblastoma, suggesting that inactivation of both RB1 alleles was critical to the development of osteosarcomas in those with inherited retinoblastoma.24, 25 Chromosome 13q LOH was also frequently observed in sporadic osteosarcomas. These molecular studies of retinoblastomas and osteosarcomas provided strong support for Knudson’s two-hit hypothesis and suggested that a variety of tumors might arise through the inactivation of various tumor suppressor loci.11, 21, 23 In addition, the studies demonstrated that the inherited and sporadic forms of a specific tumor type both appeared to arise as a result of genetic alterations in the same gene.
Cloning and analysis of the RB1 gene
The molecular cloning of the RB1 gene was facilitated by the identification in the chromosome 13q14 region of an anonymous DNA marker that detected DNA rearrangements in retinoblastomas.26 Analysis of the DNA sequences flanking this DNA marker revealed a gene with the properties expected of RB1.27–29 The RB1 gene has a complex organization, with 27 exons spanning more than 200 kb of DNA, and an RNA transcript of about 4.7 kb. The RB1 gene appears to be expressed ubiquitously rather than to be restricted to retinoblasts and osteoblasts.
Cloning of RB1 allowed study of mutations that inactivate the gene. Although gross deletions of RB1 sequences are observed in a small subset of retinoblastoma and osteosarcoma cases, most tumors appear to express full-length RB1 transcripts and lack detectable gene rearrangements when analyzed by Southern blotting.30–33 Hence, the detection of inherited and somatic mutations in the RB1 gene in most cases requires detailed characterization of its sequence. Extensive analysis of mutant RB1 alleles has provided definitive molecular evidence supporting Knudson’s two-hit model. As predicted, patients with inherited retinoblastoma have been found to have one mutated and one normal allele in their constitutional (blood) cells. In retinoblastomas of such individuals, the remaining RB1 allele has been found to be inactivated by somatic mutation, usually by loss of the normal allele through a gross chromosomal event (Figure 2), but in some cases by point mutation. Multiple tumors arising in an individual patient with inherited retinoblastoma were all found to contain the same germline mutation, but different somatic mutations affected the remaining RB1 allele. The vast majority of patients with a single retinoblastoma and no family history of the disease have two somatic mutations in their tumors and two normal alleles in their constitutional cells.
The observation that RB1 is ubiquitously expressed is rather puzzling, given the restricted spectrum of tumors that develop in patients with germline RB1 mutations. Patients with germline mutations of RB1 are at elevated risk only for the development of a rather limited number of tumor types, including retinoblastoma in childhood, osteosarcoma, soft-tissue sarcoma, and melanoma later in life. RB1 germline mutations fail to predispose to most common cancers, despite the fact that somatic RB1 mutations have been observed in a wide variety of cancer types, including breast, small cell lung, bladder, pancreas, and prostate cancers.34 It is possible that retinoblastoma protein functions differently in retinal epithelial cells than in other cell types, so that the RB1 gene acts as a “gatekeeper” in retinal cells but not in other cell types.
Function of retinoblastoma protein P105-RB
The protein product of the RB1 gene is a nuclear phosphoprotein known as p105-Rb or, more commonly, pRB. Its molecular weight is about 105,000 Da. Studies by Whyte and colleagues provided critical insights into pRB function, connecting human tumorigenesis with experimental tumors caused by DNA tumor viruses. They demonstrated that pRB formed a complex with the E1A oncoprotein encoded by the murine DNA tumor virus adenovirus type 5.35 Prior studies of E1A had established that it had many effects on cell growth, including cell immortalization and cooperation with other oncogenes (e.g., mutated ras oncogene alleles) in neoplastic transformation. It was thus hypothesized that functional inactivation of pRB through its interaction with E1A might contribute to some of E1A’s transforming functions. Additional support for that proposal was provided by data establishing that mutations inactivating the ability of E1A to bind to pRB also inactivated E1A’s transforming function.36, 37 The significance of physical interaction between pRB and a DNA tumor virus oncoprotein was further supported by the subsequent demonstration that other DNA tumor virus oncoproteins also formed complexes with pRB, including SV40 T antigen and the E7 proteins of human papillomavirus (HPV) types 16 and 18 (Figure 3).38, 39 Many of the mutations that inactivated the transforming activities of these oncoproteins also inactivated their ability to interact with pRB. Furthermore, E7 proteins from “high-risk” HPVs (i.e., those linked to cancer development), such as HPV 16 and 18, formed complexes more tightly with pRB than did E7 proteins of “low-risk” viruses (e.g., HPV types 6 and 11). These studies of pRB provided compelling evidence that DNA tumor viruses might transform cells at least in part by inactivating tumor suppressor gene products. In addition, given the critical dependence of DNA tumor viruses on harnessing the cell’s machinery for replication of the viral genome, the studies also provided support for the hypothesis that pRB might control normal cell growth by interacting with cellular proteins that regulate the cell’s decision to enter into the DNA synthesis (S) phase of the cell cycle.
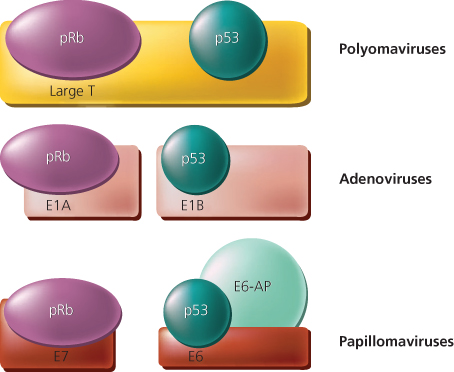
Figure 3 Schematic representation of interactions between tumor suppressor gene products and proteins encoded by DNA tumor viruses. Large T antigen from polyomaviruses [such as simian virus 40 (SV40)] bind both the retinoblastoma (pRB) and p53 proteins. For the adenoviruses and the high-risk human papillomaviruses (HPV types 16 and 18), various viral protein products complex with pRB and TP53. A cellular protein known as E6-associated protein (E6-AP) cooperates with the HPV E6 protein to complex and degrade TP53.
The functional activity of pRB is regulated by phosphorylation during normal progression through the cell cycle. Accordingly, pRB appears to be predominantly unphosphorylated or hypophosphorylated in the G1 phase of the cell cycle and maximally phosphorylated in G2 (Figure 4). The critical phosphorylation events regulating the function of pRB are likely to be mediated at the boundary between the G1 and S phases of the cell cycle by cyclin and cyclin-dependent kinase (Cdk) protein complexes.34, 40 When it is not phosphorylated, pRB forms complexes with proteins in the E2F family and inhibits transcription by recruiting proteins involved in transcriptional repression.40 When phosphorylated, pRB can no longer efficiently form complexes with E2Fs (Figure 4). The E2F proteins, when dimerized with their differentiation-regulated transcription factor partner (DP) proteins, are then capable of activating the expression of a number of genes that are likely to regulate or promote entry into S-phase, including DNA polymerase a, thymidylate synthase, ribonucleotide reductase, cyclin E, and dihydrofolate reductase.40 The E2F family members that directly affect cellular proliferation were shown in conditional mouse knockout models.41 Several other cellular proteins that bind to pRB have been identified, but their functions and the significance of their interactions with pRB remain less-well characterized than the interactions of pRB with E2Fs. Future studies will undoubtedly shed further light on the means by which loss of pRB function and that of pRB homologs p107 and p13042 contribute to cancer development.
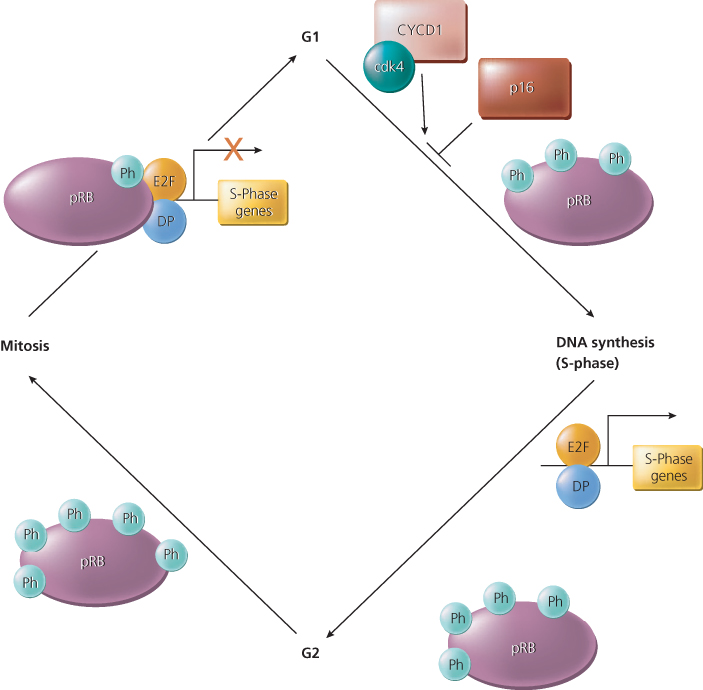
Figure 4 The function of the retinoblastoma protein (pRB) is regulated during the cell cycle by phosphorylation. The pRB protein is hypophosphorylated in the G1 phase of the cell cycle, and phosphorylation (Ph) of specific sites appears to increase during progression through the cell cycle. A protein complex that appears to phosphorylate pRB before DNA synthesis (S-phase) includes a cyclin (Cyc) and a cyclin-dependent kinase (Cdk)—for example, cyclin D1 and Cdk4. The CycD1/Cdk4 complex is regulated by the p16 inhibitor protein, which is itself the product of a tumor suppressor gene on chromosome 9p, known as CDKN2 (see text). In its hypophosphorylated state, pRB binds to E2F transcriptional regulatory proteins. When pRB is brought to the promoter regions of genes via its interaction with E2F proteins, pRB represses the expression of the E2F/DP target genes. Phosphorylation of pRB releases it from the E2F/DP protein complex and results in gene activation, including those genes involved in DNA synthesis. The figure also indicates that pRB phosphorylation increases in G2 with pRB dephosphorylated at or near anaphase.
TP53 gene
Studies in the late 1970s revealed that a cellular phosphoprotein with a relative molecular mass of about 53,000 Da formed a tight complex with SV40 T antigen; hence, the name of the p53 protein.43–45 Further work established that TP53 also formed a complex with other viral oncogene products, including the adenovirus E1B protein, and that TP53 was present at low levels in normal cells and high levels in many tumors and tumor cell lines.43, 46–48 These initial findings suggested that increased levels of TP53 might contribute to cancer. Consistent with that notion, gene transfer studies provided data demonstrating that TP53 functioned as an oncogene in in vitro experiments.47, 49–51 However, subsequent studies in human tumors showed that TP53 was in reality a tumor suppressor gene.52
The rationale for the human cancer studies was the observation that chromosome 17p LOH was common in a number of tumor types, including colorectal, bladder, breast, and lung cancer.53, 54 Detailed mapping showed that region 17p, which was lost in colorectal cancers, included the TP53 gene.52 Analysis of the sequence of the TP53 alleles retained in those cancers with 17p LOH demonstrated that the remaining TP53 allele was mutated,52 in perfect accord with Knudson’s hypothesis for the alterations expected in tumor suppressor genes. These observations were soon extended to other cancer types, and they explained many previous observations concerning TP53 that had been confusing when TP53 was believed to be an oncogene.55–59 Additional evidence that TP53 functions as a tumor suppressor gene in human cancer is provided by gene transfer studies, but such overexpression studies cannot be easily interpreted, as many genes with no role in neoplasia can inhibit the growth of transfected cells.60–63 Based on genome-wide sequencing of multiple tumor types, it is now clear that TP53 is believed to be the most frequently mutated gene in human cancers in general.64
Germline mutations in the TP53 gene have been seen in those affected by Li–Fraumeni syndrome (LFS) and in a small subset of pediatric patients with sarcomas or osteosarcomas that do not meet the more strict criteria for diagnosis of LFS.65–67 Those with LFS are at risk for developing a number of tumors, including soft-tissue sarcoma, osteosarcoma, brain tumors, breast cancer, and leukemia. Between one-half and two-thirds of patients with LFS have germline mutations in the central core domain of the TP53 coding sequences that resemble the somatic mutations frequently seen in sporadic cancers.68 Some LFS patients and families with phenotypic features of LFS have germline mutations in a gene termed hCHK2 that phosphorylates TP53 and controls the cell’s response to DNA-damaging events.69
In addition to somatic and inherited mutations in the gene, TP53 function can be inactivated by other mechanisms. As noted earlier, most cervical cancers contain high-risk or cancer-associated HPV genomes (i.e., HPV type 16 or 18). The E6 gene product of high-risk, but not low-risk, HPV types binds to a cellular protein known as E6-AP (for E6-associated protein) and stimulates TP53 degradation.70–74 A cellular TP53-binding protein known as mouse double minute 2 (MDM2) is overexpressed in a subset of soft-tissue sarcomas as a result of gene amplification involving chromosome 12q sequences.75 More recent studies have identified another TP53-binding protein, MDM4, which is overexpressed in a variety of cancers as a result of gene amplification of chromosome 1q sequences.76 DNA transfection studies have shown that both the MDM2 and MDM4 genes can function as oncogenes when overexpressed. The oncogenic function is presumably mediated through their binding to and inactivation of TP53. Both proteins mask TP53’s transcriptional activation domain and promote TP53’s ubiquitination and subsequent degradation by the proteasome.77–79 Consistent with the notion that MDM2 is a critical inhibitor of TP53 function, sarcomas with MDM2 amplification and overexpression rarely harbor somatic mutations in TP53.80 Disruption of the MDM2 and MDM4 genes in the germline of mice is lethal, probably because such disruption allows unregulated activity of TP53. Accordingly, disruption of the murine TP53 gene rescues MDM2-deficient and MDM4-deficient mice from embryonic lethality.81, 82 Other mechanisms of regulating TP53 function have also been described, including mutation of a nuclear-cytoplasmic shuttle protein called nucleophosmin in almost 100% of adult acute myelogenous leukemias that demonstrate cytoplasmic localization of this protein, with the notable exception of acute promyelocytic leukemia.83
TP53 function
The p53 protein has been shown to function as a transcriptional regulatory protein84, 85 though other, nontranscriptional functions for p53 have been described (ref. to recent review). In its wild-type state, the p53 protein is capable of binding to specific DNA sequences with its central core domain (Figure 5). The amino terminal sequences of p53 function as a transcriptional activation domain, and the carboxyl terminal sequences appear to be required for TP53 to form dimers and tetramers with itself. TP53 activates transcription of a number of genes with roles in the control of the cell cycle, including WAF1/CIP1/p21 (which encodes a regulator of Cdk activity),86 MDM2 (as noted earlier, encoding a protein that is a known negative regulator of TP53), and 14-3-3 (a regulator of G2/M progression),87 and various genes that likely function in apoptosis, including PUMA and NOXA. Experimental disruption of these genes by targeted homologous recombination can re-create some of the phenotypes associated with TP53 inactivation.88, 89
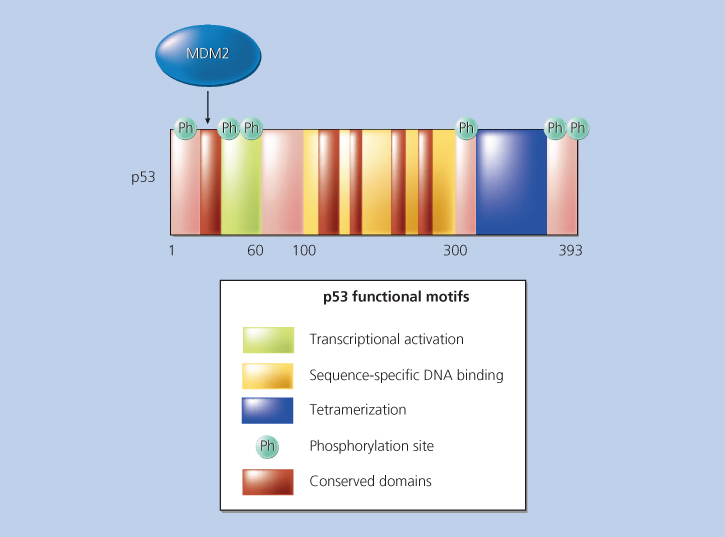
Figure 5 TP53 functional motifs. Sequences of TP53 involved in transcriptional activation, sequence-specific DNA binding, tetramerization, and binding by the MDM2 protein are indicated. The five distinct regions of TP53 sequence that are highly conserved between p53 proteins of diverse species are indicated. In addition, the locations of several sites in the protein that are phosphorylated (Ph) and that regulate TP53 function are indicated.
The vast majority of the somatic mutations in TP53 are missense mutations leading to amino acid substitutions in the central portion of the protein (exons 5–9).90 Consistent with the structure of the p53 protein, these missense mutations have marked effects on the p53 protein’s ability to bind to its cognate DNA recognition sequence through either of two mechanisms.91 Some mutations (e.g., mutations at codons 248 or 273) alter TP53 sequences that are directly responsible for sequence-specific DNA binding. Other mutations (e.g., codon 175) appear to affect the folding of TP53 and thus indirectly affect its ability to bind to DNA. Evidence that these missense mutations can confer “gain of function” rather than a dominant negative effect was demonstrated via “knock in” mouse models, whereby precise missense mutations were introduced into the endogenous TP53 gene.92–96
Under some circumstances, TP53 acts at the G1/S checkpoint to regulate the cell’s decision to synthesize DNA, although TP53 also appears to have a critical function at G2/M.97, 98 In other settings, TP53 appears to exert control over the cell’s decision to undergo apoptosis or programmed cell death.85 Of particular interest with regard to cancer treatment are data suggesting that some tumor cells lacking TP53 function are less sensitive to irradiation and chemotherapeutic agents such as cisplatin.92, 99, 100 However, studies of other tumor cells suggest that TP53 status shows a very different relationship to chemotherapeutic response, with cells that lack functional TP53 being markedly sensitive to DNA-damaging agents but resistant to 5-fluorouracil.101 Thus far, studies of primary human cancers have emphasized that a rather complex relationship is likely to exist between TP53 mutational status and the responsiveness of cancer cells to chemotherapy or radiation therapy, or both. In particular, it is difficult to distinguish the effects of TP53 mutation on the natural progression of disease from its effects on responses to treatment and other cellular stresses.102, 103 Hopefully, further research on TP53 will clarify our understanding of its normal functions, the basis for its frequent inactivation in many different cancers, and the consequences of its inactivation on tumor growth and response to therapy.
Cyclin-dependent kinase inhibitor 2A locus
Studies of the cyclin-dependent kinase inhibitor 2A (CDKN2A) locus on chromosome 9p illustrate well how observations from initially disparate lines of investigation often converge to implicate a particular locus as a critical factor in cancer development. The LOH of chromosome 9p was frequently found in many different tumor types, including melanoma, glioma, nonsmall cell lung, bladder, head and neck cancers, and leukemia.104–107 Of considerable interest were observations establishing that a subset of such tumors had homozygous (complete) deletions affecting the 9p21 region,108–110 strongly supporting the existence of a tumor suppressor gene in the region. In addition to the frequent somatic alterations of chromosome 9p sequences in cancers, linkage studies of some families with inherited melanoma indicated a melanoma predisposition gene mapped to essentially the same region of 9p.111 These observations stimulated great interest in the chromosome 9p region presumed to contain the tumor suppressor gene or genes. One of the genes identified in the region as a result of positional cloning efforts was initially termed multiple tumor suppressor 1 (MTS1).112 Sequence analysis of MTS1 showed that it was identical to a previously described gene that encoded the Cdk inhibitor protein known as p16.113 Because the p16 protein functioned by inhibiting Cdk4 and Cdk6, it was termed an inhibitor of cyclin-dependent kinase 4 (INK4) protein. Another highly related gene, mapping immediately next to the p16/MTS1 gene on chromosome 9p, was found to encode a second INK4 protein, known as p15 (Figure 6). The gene encoding the p16 protein is most often termed INK4A, and the gene for p15 is INK4B.114, 115 The approved symbols are CDKN2A and CDKN2B, respectively.
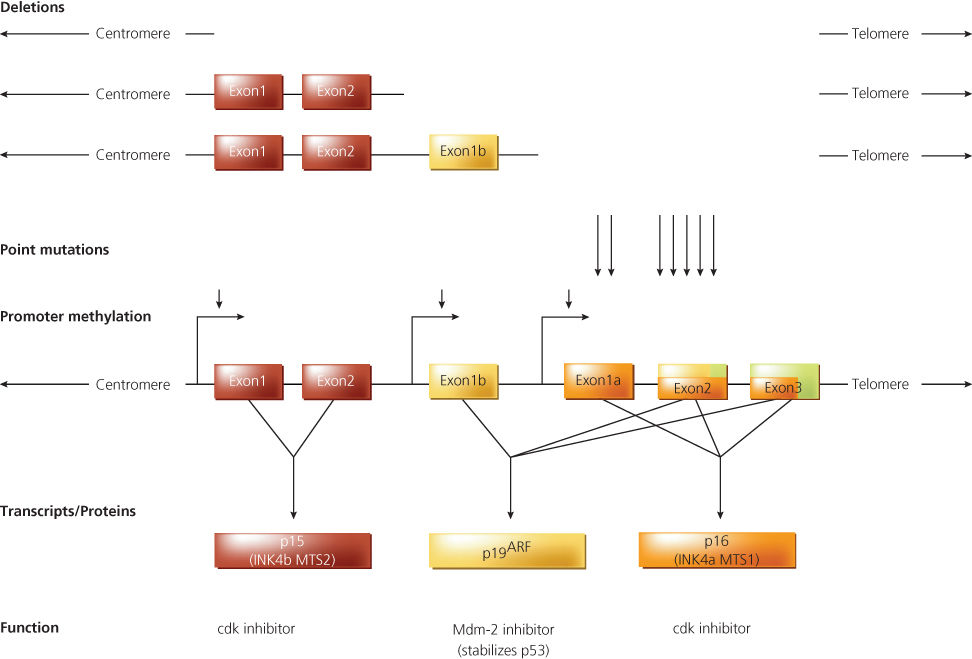
Figure 6 Genomic structure, mutations, and transcripts of the CDKN2B (p15) and CDKN2A (p16/p19ARF) locus. The origins of the p15, p16, and p19ARF transcripts are shown schematically, along with a representative depiction of genomic deletions, point mutations (arrows), and promoter methylation (arrowheads) noted in human cancers. The exons of the CDKN2B and CDKN2A loci are shown as rectangles. The transcripts/proteins and presumed functions of the transcripts/proteins are indicated. The red rectangles indicate the open reading frame in transcripts encoding p15; the yellow rectangles indicate the open reading frame present in transcripts encoding p19ARF; and the lavender rectangles indicate the open reading frame present in transcripts encoding p16. The size of the locus, exons, and transcripts are not shown to scale.
Subsequent studies show that heterozygous mutations in CDKN2A are present in some families with inherited predispositions to melanoma or pancreatic cancer.116–119 Somatic mutations in CDKN2A are present in many different cancer types, including but not limited to melanoma, glioma, pancreatic and bladder cancers, and leukemia. In some tumors, deletions affecting the CDKN2A gene also involve the CDKN2B gene. In rare tumors, deletions inactivate CDKN2B but not CDKN2A.120 The prevalence and specific nature of CDKN2A mutations vary markedly from one tumor type to another. In contrast to other tumor suppressor genes, such as RB1 and TP53, homozygous deletion is a fairly common mechanism of CDKN2A inactivation in cancer.121
Detailed studies of the CDKN2A locus led to the identification of a novel alternative transcript containing nucleotide sequences identical to those in transcripts for the p16INK4A protein, but with unique 5′ sequences (Figure 6).114, 115, 122 The alternative CDKN2A locus transcript encodes a protein known as p14 alternative reading frame (p14ARF). Of note, human p14ARF is the same product as the originally identified mouse protein p19ARF, which was designated as p19 due to its apparent weight.
The p14ARF protein contains sequences from a distinct first exon (exon 1β). Exon 1β is located upstream of exon 1α, the first exon present in transcripts for p16 (Figure 6). Exon 1β is spliced to exon 2, which, along with exon 3, is present in the transcripts for both the p14ARF and p16INK4A proteins. However, the p14ARF protein shares no sequence similarity with the p16INK4A protein because p14ARF synthesis initiates at a unique methionine codon in exon 1β and continues through exon 2, using an alternative open reading frame with no similarity to the p16INK4A open reading frame. Careful studies of somatic and inherited mutations at the CDKN2A locus indicate that localized mutations inactivating the p16INK4A protein are common in human cancer, with more than 60 germline mutations identified, but that localized mutations inactivating p14ARF are uncommon.114, 115 However, the frequent occurrence of homozygous deletions at the CDKN2A locus implies that mutational inactivation of both proteins—and of p15INK4B—may be strongly selected for during tumor development (Figure 6). Other findings suggest that p16INK4A and p14ARF expression may be lost in some tumor types as a result of methylation of DNA regulatory sequences at the CDKN2A locus (Figure 6).123–125 Recent studies have demonstrated the importance of DNA methyltransferases in initiating and maintaining epigenetic silencing of the p16 tumor suppressor.126, 127 Furthermore, studies of mice with germline inactivation of p14ARF and p16INK4A indicate that these proteins function as tumor suppressor genes in vivo.128–130
The mechanism through which the p16INK4A protein controls tumorigenic growth is apparently through its inhibition of Cdk4 activity. As indicated earlier, phosphorylation of pRB impedes its ability to transcriptionally regulate E2F-target genes (Figure 4). The cyclin D1/Cdk4 complex has a critical role in regulating pRB phosphorylation and function.124 Hence, the p16INK4A protein, by virtue of its regulation of Cdk4 activity, is, in turn, a critical factor in regulating pRB phosphorylation. Presumably, inactivation of p16INK4A results in inappropriate phosphorylation of pRB and a subsequent inability of hyperphosphorylated pRB to bind E2Fs and appropriately regulate gene expression at the G1–S transition. p14ARF functions as a tumor suppressor by direct binding to the MDM2 protein, inhibiting the MDM2-induced degradation of TP53, thus maintaining appropriate function of TP53 in cells,115 much like p16INK4A maintains normal pRB function.
Contrary to this interpretation, however, is the fact that alterations of p14ARF and TP53 often coexist in cancer cells, suggesting that they do not alter the same pathway, whereas Rb and p16INK4A alterations are mutually exclusive, supportive of the fact that they affect the same pathway.131 Nevertheless, these findings emphasize the concept that oncogenes and tumor suppressor genes do not function in isolation. Rather, they function in intricately linked cascades or networks that have important consequences for both tumorigenesis and therapy (Figure 7).85, 132
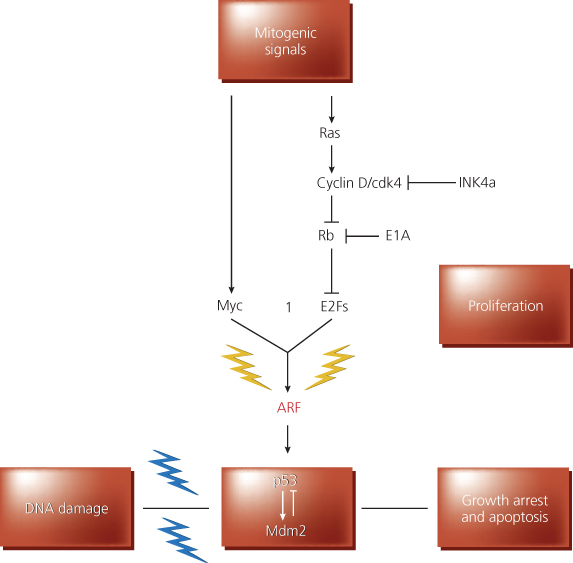
Figure 7 Role of the p19*RF protein in checkpoint control. The p19ARF protein (ARF) responds to proliferative signals normally required for cell proliferation. When these signals exceed a critical threshold, the ARF-dependent checkpoint (yellow lightning bolts) is activated, and ARF triggers a TP53-dependent response that induces growth arrest or apoptosis, or both. Signals now known to induce signaling via the ARF-TP53 pathway include Myc, E1A, and E2F-1. In principle, “upstream” oncoproteins, such as products of mutated ras alleles, constitutively activated receptors, or cytoplasmic signal transducing oncoproteins, might also trigger ARF activity via the cyclin D-Cdk4-RBE2F or Myc-dependent pathways, both of which are normally necessary for S-phase entry. In inhibiting cyclin D-dependent kinases, p16INK4A can dampen the activity of mitogenic signals. In the figure, E1A is shown to work, at least in part, by opposing RB function. For simplicity, Myc and E2F-1 are shown to activate only TP53 via the effects on ARF, though highly overexpressed levels of these proteins can activate TP53 in ARF-negative cells, albeit with an attenuated efficiency. ARF activation of TP53 likely depends on inactivation of Mdm2-specific function(s). DNA damage signals (e.g., ionizing and ultraviolet radiation, hypoxic stress) activate (blue lightning bolts) TP53 through multiple signaling pathways.
Adenomatous polyposis coli gene
Identification and germline mutations
Colon cancers arising in a minority of hereditary syndromes do so from a state of polyposis. One of the polyposis syndromes is known as familial adenomatous polyposis (FAP) or adenomatous polyposis coli (APC). FAP is an autosomal dominant disorder affecting about 1 in 8000 individuals in the United States. The syndrome is characterized by the development of hundreds of adenomatous polyps in the colon and rectum of affected individuals by early adulthood. The lifetime risk of colorectal cancer in those with the classic form of FAP is extremely high, approaching 100% by age 60 years.
An interstitial deletion of chromosome 5q in a patient with features of FAP, but without any family history of the syndrome, greatly aided localization of the APC gene.133 Subsequent DNA linkage studies confirmed that, in multiple kindreds with FAP, or the related condition known as Gardner syndrome, the polyposis phenotype segregated with DNA markers near 5q21.134, 135 In 1991, positional cloning efforts identified the APC gene as the specific gene responsible for FAP.136–139 The APC gene is large, with more than 15 exons, and alternative splicing affects the 5′ untranslated portion of transcripts. The predominant APC transcript encodes a 2843-amino acid protein expressed in many adult tissues.
In the great majority of individuals with FAP, heterozygous germline mutations can be identified in the APC gene.140–142 All of the germline APC mutations in those with FAP appear to inactivate APC protein function. The overwhelming majority of these germline mutations are localized nonsense or frameshift mutations in the 5′ half of the coding region of APC (Figure 8). Consistent with Knudson’s two-hit hypothesis, inactivation of the remaining wild-type APC allele by somatic mutation in those carrying a germline APC mutation is observed in the cancers that arise.143, 144 Correlations between the location of a particular germline APC mutation and clinical features have been found,145 although clear insights into the molecular basis for the predisposition to extracolonic tumors (e.g., jaw osteomas and desmoid tumors) in FAP patients with variant phenotypes are lacking. However, some light has been shed on the variability in polyp number seen in families with polyposis.143, 146 Mutations in the 5′ region of the APC gene appear to be correlated with an attenuated phenotype attributable to reentry of the ribosome on the APC transcript downstream of the premature stop codon, resulting in an APC protein that retains some of its normal activity.146 Mutations in 3′ third of the APC gene are also associated with a milder polyposis phenotype than mutations in the central third of the gene, perhaps because the mutated APC proteins similarly retain some tumor suppressor activity. Unexpectedly, extracolonic features such as desmoid tumors appear to be more common in patients with 3′ mutations.143 Finally, a missense mutation in the middle of the APC gene has been found to predispose to colorectal cancers in Ashkenazi Jewish families.147 This mutation does not alter the function of the gene product, but creates a “hot spot” that appears to be highly mutable, resulting in somatic deletions or insertions of surrounding nucleotides that produce truncations.
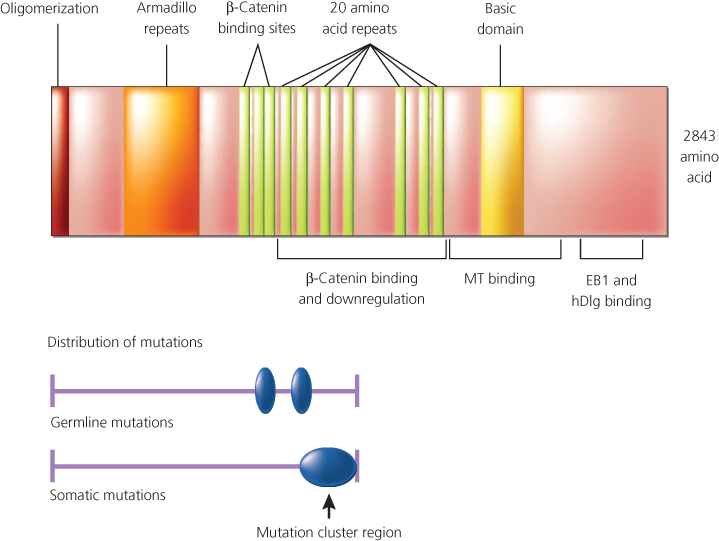
Figure 8 Schematic representation of Apc protein domains with respect to mutational analysis results. The relative positions of various Apc domains. A putative domain involved in homo-oligomerization of Apc is located at the amino-terminus. Also noted are a series of repeats of unknown function with similarity to the Drosophila armadillo protein, sequences known to mediate binding to p-catenin and its downregulation, a basic domain in the carboxy-terminal third of the protein that appears to facilitate complexing with microtubules (MT), and sequences near the carboxy terminus of Apc that are known to interact with the EB1 and human homolog of the Drosophila disc large (hDlg) protein. Germline mutations in the APC gene (predominantly chain terminating) are dispersed throughout the 5′ half of the sequence, with two apparent “hot spots” at codons 1061 and 1309. Somatic mutations in the APC gene in colorectal cancer appear to cluster in a region termed the “mutation cluster region,” and mutations at codons 1309 and 1450 are most common.
Somatic mutations in sporadic colon tumors
Although germline APC mutations are an uncommon cause of colorectal cancer in the general population and are present in only about 0.5% of all colon cancers, somatic APC mutations are present in the vast majority of sporadic colorectal adenomas and carcinomas.148 The initial observation suggesting that APC inactivation might be common in colon tumors was the observation that the chromosome 5q region containing the APC gene was affected by LOH in many sporadic colorectal adenomas and carcinomas.54, 149 Since the identification of the APC gene, detailed analyses of the somatic mutations inactivating the APC gene in colorectal tumors have been carried out. The somatic APC mutations in sporadic tumors are similar in nature and location to the germline APC mutations found in those with FAP (Figure 8). Present findings suggest that up to 90% of colorectal tumors, regardless of their size or particular histopathologic features, harbor somatic mutations that inactivate APC. 143–150
Function
The APC gene encodes a large protein of roughly 300 kDa that is hypothesized to regulate cell adhesion, cell migration, or apoptosis in the colonic crypt. The localization of the APC protein in the basolateral membrane of colonic epithelial cells, with an apparent increase in APC expression in cells near the top of the crypt, implies that APC may regulate shedding or apoptosis of cells as they reach the crypt apex.151 Perhaps consistent with this view, restoration of APC protein expression in colorectal cancer cells lacking endogenous APC expression has been reported to promote apoptosis.152, 153
The APC protein binds to a number of proteins, including β-catenin, Y-catenin (also known as plakoglobin), glycogen synthase kinase 3β (GSK3β), end-binding protein 1 (EB1), human Drosophila large discs (hDLG), microtubules, and the related proteins axin and conductin.154 With the exception of β-catenin, GSK3β, and the conductin and axin proteins, the significance and role of APC’s interactions with its various binding partners are not well understood. Several lines of evidence imply that APC has a critical function in regulating β-catenin.154, 155 β-Catenin is an abundant cellular protein, first identified because of its role in linking the cytoplasmic domain of the E-cadherin cell–cell adhesion molecule to the cortical actin cytoskeleton, via β-catenin’s binding to α-catenin. The truncated (mutant) APC proteins present in many colorectal cancers lack some or all of the repeat motifs crucial for binding to β-catenin. APC not only binds to β-catenin, but, in collaboration with the GSK3β enzyme and other proteins, such as axin and conductin, it appears to regulate the abundance of β-catenin in the cytosol via phosphorylation. In colorectal cancers in which APC is mutated and unable to bind or effectively coordinate the regulation of β-catenin, β-catenin accumulates in the cell, complexes with the transcription factor T-cell factor-4 (Tcf-4), and translocates to the nucleus (Figure 9). Once there, β-catenin functions as a transcriptional coactivator, activating expression of Tcf-4-regulated genes, typically bypassing regulatory proteins, such as Wnt. Consistent with the notion that β-catenin is a critical target of APC regulation, somatic mutations in β-catenin have been found in the small fraction of colorectal cancers lacking APC mutations.156–158 These mutations consistently alter GSK3β phosphorylation consensus sites near the amino terminus of the β-catenin protein, and the mutations presumably render the defective β-catenin proteins oncogenic as a result of their resistance to degradation by APC and GSK3β. Although somatic mutations in APC appear to be rare in cancers arising outside the colon and rectum, oncogenic mutations in β-catenin’s N-terminus have been observed in many different cancer types.159, 160
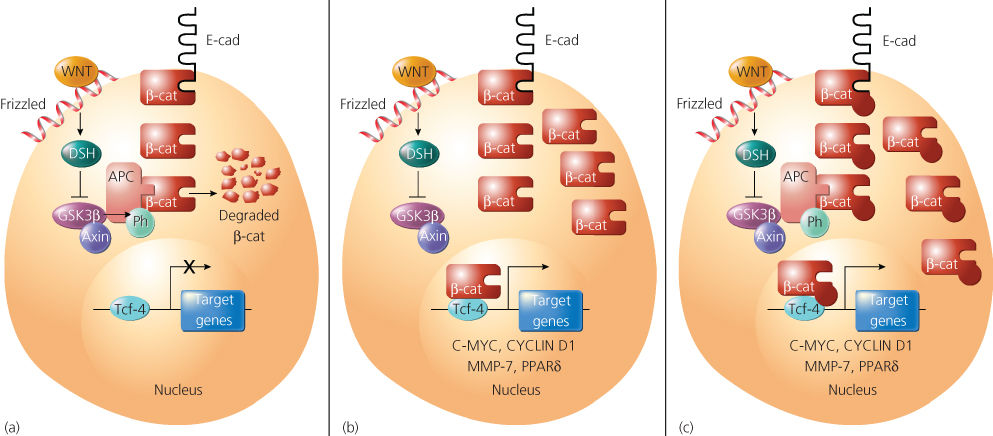
Figure 9 A model indicating the function of the Apc, axin, and Gsk3p proteins in the regulation of p-catenin (p-cat) in normal cells, and the consequence of Apc or p-cat defects in cancer cells. p-Cat is an abundant cellular protein, and much of it is often bound to the cytoplasmic domain of the E-cadherin (E-cad) cell–cell adhesion protein. (a) In normal cells, the proteins glycogen synthase kinase 3p (Gsk3p), Apc, and axin function to promote degradation of free cytosolic p-cat, probably as a result of phosphorylation of the N-terminal sequences of p-cat by Gsk3p. Gsk3p activity and p-cat degradation are inhibited by activation of the wingless (Wnt) pathway, as a result of the action of the Frizzled receptor and disheveled (Dsh) signaling protein. (b) Mutation of Apc in colorectal and other cancer cells results in accumulation of p-cat, binding to Tcf-4, and transcriptional activation of Tcf-4 target genes, such as c-myc, cyclin D1, MMP-7, and PPARS (see text). (c) Point mutations and small deletions in p-cat in cancer cells inhibit phosphorylation and degradation of p-cat by Gsk3p and Apc, with resultant activation of c-myc and other Tcf-4 target genes.
Wilms tumor gene
Wilms tumors are the most common renal neoplasm in children, accounting for approximately 6% of all pediatric cancers.161 Wilms tumors are similar to retinoblastomas in a number of ways: both can occur bilaterally or unilaterally, with single or multiple foci, and in a sporadic or inherited fashion. The two mutation model originally proposed for retinoblastoma was also proposed to explain Wilms tumor.162 However, hereditary cases are not as common among Wilms tumor patients as they are in retinoblastoma patients. Almost all patients inheriting a mutation at the RB1 locus develop a retinoblastoma, but only approximately 50% of individuals carrying a germline mutation predisposing to Wilms tumor develop the disease (i.e., lower penetrance).161
Perhaps the first finding to offer insight into an inherited genetic basis for Wilms tumor was a report describing six patients with Wilms tumor and sporadic aniridia (i.e., congenital absence of the iris).163 It was proposed that the simultaneous occurrence of these two very rare conditions might result from chromosomal aberrations affecting two or more loci, a situation now often called a contiguous gene syndrome—mutation of one locus presumably leading to aniridia and mutation of another leading to Wilms tumor. This hypothesis was subsequently supported by the discovery of interstitial deletions of chromosome 11p13 in peripheral blood samples from children with the WAGR syndrome (Wilms tumor with aniridia, genitourinary abnormalities, and mental retardation) of Wilms tumor.164 Cytogenetic studies of tumor tissues in a few cases of sporadic-type Wilms tumors revealed deletions or translocations of chromosome band 11p13.165, 166 Subsequent studies of paired samples of Wilms tumor and normal cells from patients, using probes that detect restriction fragment length polymorphisms (RFLPs) on chromosome 11p, revealed that LOH of 11p occurred frequently in Wilms tumors of both the inherited and sporadic types.167–170
The Wilms tumor 1 (WT1) gene was identified in 1990 by virtue of mutations inactivating the gene in patients with the WAGR syndrome and by analysis of somatic mutations in the gene in tumors from a minority of patients with unilateral Wilms tumor and no associated congenital malformation.171 WT1 is encoded by 10 exons and its transcripts are subject to alternative splicing.172, 173 In contrast to the rather ubiquitous expression of the RB1, TP53, and APC genes, expression of the WT1 gene appears to be restricted to embryonic kidney and a small subset of other tissues.174, 175 WT1 messenger ribonucleic acids (mRNAs) encode proteins with molecular masses of 45,000–49,000 Da and 4 zinc-finger motifs. Based on their predicted amino acid sequences, the WT1 proteins were suspected from the outset to function in transcriptional regulation.174 Several studies provide evidence to support that notion, although some WT1 isoforms may have a role in RNA processing, rather than in transcription regulation.173, 174 WT1 proteins suppress the transcriptional activity of promoter elements from a number of growth-inducing genes, including the genes for early growth response (EGR1), insulin-like growth factor-2 (IGF-2), and platelet-derived growth factor A (PDGFA) chain, suggesting that WT1 may function in gene repression.176 Other studies suggest that WT1 can activate or repress gene expression, depending on the cell type and promoter context.177 Consistent with the notion that WT1 may have a physiologic function in transcriptional activation, recent work indicates that WT1 activates expression of amphiregulin, a member of the epidermal growth factor family.178 Loss of amphiregulin expression may contribute to loss of appropriate differentiation during Wilms tumor development. Adding to the complex nature of WT1’s role as a transcriptional regulator, recent studies suggest that certain WT1 splice variants have dramatically different effects in their ability to regulate gene expression,179, 180 including acting as an oncogene in a variety of adult cancers.
WT1 inactivation clearly contributes to Wilms tumor development in those with the WAGR syndrome. Moreover, approximately 10% of apparently sporadic Wilms tumors harbor detectable somatic mutations in the WT1 gene.181 Nevertheless, much evidence indicates that Wilms tumors arise through mutations in genes other than WT1. First, the chromosome 11p allelic losses seen in Wilms tumor frequently involve band 11p15, but not band 11p13, where the WT1 gene resides.181–183 Second, the 11p15 region harbors a gene responsible for Beckwith–Wiedemann syndrome (BWS), a congenital disorder in which affected individuals manifest hyperplasia of the kidneys, endocrine pancreas, and other internal organs; macroglossia; and hemihypertrophy.184, 185
Those affected by BWS are also at increased risk of developing embryonic tumors, such as hepatoblastoma and Wilms tumor. Finally, linkage studies of three families with dominant inheritance of Wilms tumor exclude linkage of the susceptibility locus in those families to any part of chromosome 11p.186, 187 These data and others suggest that germline mutations in any one of at least three different genes (i.e., WT1, the BWS gene, and at least one gene not on chromosome 11p reference to haber’s paper on X-chromosome gene) predispose to Wilms tumor. Whether a combination of inherited and somatic mutations in more than one of those genes is ultimately required for the transformation of a developing kidney cell into a Wilms tumor, or whether alternative genetic pathways for the development of Wilms tumors exist, remains to be established. The genetic heterogeneity observed among Wilms tumors provides an important contrast to the apparently less complex genetic pathway of retinoblastoma.
Neurofibromatosis 1 and 2 genes
Neurofibromatosis 1 gene
Von Recklinghausen’s disease, also called neurofibromatosis 1 (NF1), is a dominantly inherited syndrome with variable disease manifestations. The consistent feature is that tissues derived from the neural crest are commonly affected. In addition to the nearly uniform development of neurofibromas, NF1 patients are at elevated risk for developing pheochromocytomas, schwannomas, neurofibrosarcomas, and primary brain tumors.188–190 The NF1 gene was initially localized to the pericentromeric region of chromosome 17q by linkage analyses.191, 192 Subsequently, karyotype studies of two NF1 patients identified germline chromosomal rearrangements involving band 17q11.193, 194 In further work, both patients were found to have genetic alterations of a localized region of band 17q11. Intensive positional cloning efforts in this chromosome region led to the identification of the NF1 gene in 1991.195–197 The NF1 gene is large, spanning roughly 350 kb of DNA, and it encodes a protein product with a molecular mass of about 300 kDa.188, 190, 198 Although germline mutations in the NF1 gene are believed to underlie the development of the associated disease features in all or nearly all NF1 patients, specific germline NF1 mutations have been identified in approximately one-half to two-thirds of NF1 patients. 188, 190, 198, 199 Difficulties in identifying germline mutations in the NF1 gene in the remaining NF1 patients may be a result of the inherent inefficiencies and insensitivity associated with mutation detection strategies in such a large gene, and over 1500 distinct mutations have thus far been described.
In addition to germline NF1 mutations in those patients with NF1, the NF1 gene is affected by somatic mutations in a fraction of colon cancers, melanomas, neuroblastomas, and bone marrow cells from patients with the myelodysplastic syndrome.188, 198, 200–202 Consistent with its presumed tumor suppressor role, the mutations inactivate NF1. Studies of leukemias arising in pediatric neurofibromatosis patients provide the clearest evidence that both copies of the NF1 gene are inactivated during tumorigenesis,203 as predicted by the Knudson model. Like the RB1, TP53, and APC genes, the NF1 gene is expressed ubiquitously. Thus, as for other inherited cancer syndromes, the basis for the tissue specificity of the malignant tumors observed in neurofibromatosis patients is puzzling. The NF1 protein product, termed neurofibromin, is a member of the guanosine triphosphate (GTPase)-activating protein family.188, 204–206 Perhaps the best studied GAP is Ras-GAP, which markedly enhances the GTPase activity of the wild-type K-Ras, H-Ras, and N-Ras proteins. Although the means through which NF1 defects alter cell growth is not fully described, it is currently postulated that inactivation of neurofibromin function leads to alterations in signaling pathways regulated by small Ras-like GTPase proteins.207
Neurofibromatosis 2 gene
Neurofibromatosis 2 (NF2—also known as central neurofibromatosis) is an autosomal dominant disorder that is distinct from NF1 in both genetic and clinical features.188, 208, 209
A hallmark of NF2 is the occurrence of bilateral schwannomas that affect the vestibular branch of the eighth cranial nerve (acoustic neuromas). NF2 patients are also at elevated risk for meningiomas, spinal schwannomas, and ependymomas. The neurofibromatosis 2 (NF2) gene was mapped to chromosome 22q by a combination of linkage analyses and LOH studies210–212 and was cloned in 1993 using positional cloning approaches.213, 214 Germline mutations inactivating the NF2 gene were observed in those patients with NF2, and somatic NF2 mutations were also observed in a subset of sporadic schwannomas and meningiomas. Somatic NF2 mutations in most other tumor types appear to be infrequent. However, preliminary studies indicate that the NF2 gene may be frequently affected by somatic mutations in malignant mesotheliomas215, 216 despite this tumor type not being seen at increased frequency in patients with NF2.209 The NF2 gene encodes a protein, Merlin, with a high sequence homology to a cytoskeletal protein family [ERM (Ezrin/Radixin/Moesin)], which acts as linker proteins between integral membrane proteins and scaffolding proteins of the filamentous submembrane lattice.214 Consequently, NF2 gene alterations might contribute to tumor development, at least in part, by effects on cell shape, cell–cell interactions, or cell movement (or a combination).
von Hippel–Lindau gene
von Hippel–Lindau (VHL) syndrome is a rare dominant disorder predisposing affected individuals to the development of hemangioblastomas of the central nervous system and retina, renal carcinomas of clear cell type, and pheochromocytomas.217–219 The VHL gene was mapped to chromosome 3p by linkage analysis. As with many other inherited cancer genes, LOH studies established that the VHL gene behaves as a typical tumor suppressor gene, with both alleles inactivated during tumorigenesis.218, 220 Positional cloning efforts identified the VHL gene in 1993.221 Germline mutations inactivating one VHL allele are seen in the majority of individuals in families displaying features of the VHL syndrome.217–219 As with some other inherited cancer syndromes, preliminary genotype–phenotype relationships have been observed. Specifically, a certain class of VHL germline mutations is associated with the development of renal cancer only, a second class is linked to predisposition to both renal cancer and pheochromocytoma, and yet a third mutation class is associated only with pheochromocytoma.218 Somatic mutations in the VHL gene are also seen in more than 80% of sporadic renal cell carcinomas of the clear cell type, but not in renal cell carcinomas of other histopathologic types (e.g., papillary type).218, 219 Approximately, 20% of sporadic clear cell renal cancers do not carry a detectable mutation in the VHL gene. However, in many of these cases, the VHL gene may be inactivated by epigenetic silencing,222 a mechanism noted earlier in this chapter in connection with inactivation of the CDKN2A locus. In tumor types other than clear cell renal cancer, inactivation of the VHL gene appears to be uncommon.218
The VHL gene encodes a 213-amino acid protein whose major function appears to be in the regulation of angiogenesis through protein degradation. The protein encoded by VHL is part of a ubiquitin ligase complex that degrades hypoxia-inducing factor 1a (HIF-1a) in the presence of oxygen. In the absence of oxygen in normal cells, or when VHL is mutated in tumor cells, the HIF-1a transcription factor is stabilized, leading to the expression of cytokines such as vascular endothelial growth factor and the stimulation of angiogenesis. Further biochemical and cell biology studies on VHL and renal cell carcinomas are likely to offer definitive insights into angiogenesis, one of the most important stromal processes associated with neoplasia.223, 224
Genomic stability genes
Several recessive cancer predisposition syndromes resulting from inactivation of genes that function in DNA damage recognition and repair have been described, including ataxia telangiectasia (AT), Bloom syndrome, xeroderma pigmentosum, and Fanconi anemia. In each case, the specific cancer type and the DNA-damaging agents that increase cancer risk are distinct. Although AT heterozygotes may subtly increase the risk of breast cancer,225 in other recessive cancer syndromes, only homozygotes appear to clearly increase cancer risk. This observation contrasts sharply with the picture in the dominant cancer predisposition syndromes discussed earlier (e.g., inherited retinoblastoma, FAP, NF1, and NF2), where heterozygotes have a clearly elevated cancer risk. Homozygous mutations in tumor suppressor genes rarely if ever exist, probably because the condition is lethal during embryogenesis. It is important to remember that the tumor suppressor genes do not only function as guardians against cancer; their main function is the control of normal cell balance, and their inactivation is expected to be incompatible with normal embryonic development.
Because recessive cancer syndromes are quite rare, this discussion of the role of genomic stability genes in cancer will focus on syndromes that are inherited in an autosomal dominant fashion. These syndromes include the most common types of familial cancers, predisposing to tumors of the colon, breast, and other organs.
DNA mismatch repair gene defects and hereditary nonpolyposis colorectal cancer
Familial clustering of colon cancer has long been recognized, with approximately 5% of all colon cancers attributable to inheritance of a gene defect with a strong effect on cancer risk and another 10–15% with a moderate effect on risk. Germline APC mutations are responsible for 0.5–1% of colorectal cancer cases in the Western world, and hereditary nonpolyposis colorectal cancer (HNPCC) is responsible for 2–4%.226–228
Diagnostic criteria for identifying those individuals and families most likely to be affected by HNPCC have been determined,226–229 despite the absence of overt clinical findings before cancer diagnosis, and the potential for chance clustering of colon cancer within a family. Representative diagnostic criteria include (1) exclusion of familial polyposis; (2) colorectal cancer in at least three relatives, one of them being a first-degree relative of the others; (3) two or more successive generations affected; and (4) at least one of the affected individuals being younger than 50 years of age at the time of diagnosis. Although not all individuals affected by HNPCC meet these criteria, familial aggregations of colorectal cancer that are likely to have a genetic basis distinct from that underlying the majority of HNPCC cases can be excluded. 226, 228
Several genes responsible for HNPCC have been identified, including two on chromosome 2p [mutS homologs 2 and 6 (MSH2, MSH6)], another on chromosome 3p [mutS homolog 1 (MLH1)], and another on chromosome 7p [postmeiotic segregation 2 (PMS2)]. Together, germline mutations in these four genes account for virtually all classic HNPCC cases.226, 228–231 The protein products of the MSH2 and MLH1 genes appear to have critical roles in the recognition and repair of DNA mismatches (Figure 10). In cells with one normal and one mutant allele of a DNA mismatch repair (MMR) gene, DNA repair is minimally impaired, if at all. However, inactivation of the remaining allele can occur as a result of somatic mutation in a normal epithelial cell. This “second hit” abrogates MMR function, and hundreds to thousands of mutations may thereby occur during each subsequent cell-division cycle. Because these mutations preferentially arise in mononucleotide, dinucleotide, and trinucleotide repeat tracts (i.e., microsatellite sequence tracts), the phenotype is often called the microsatellite instability (MSI) phenotype.226
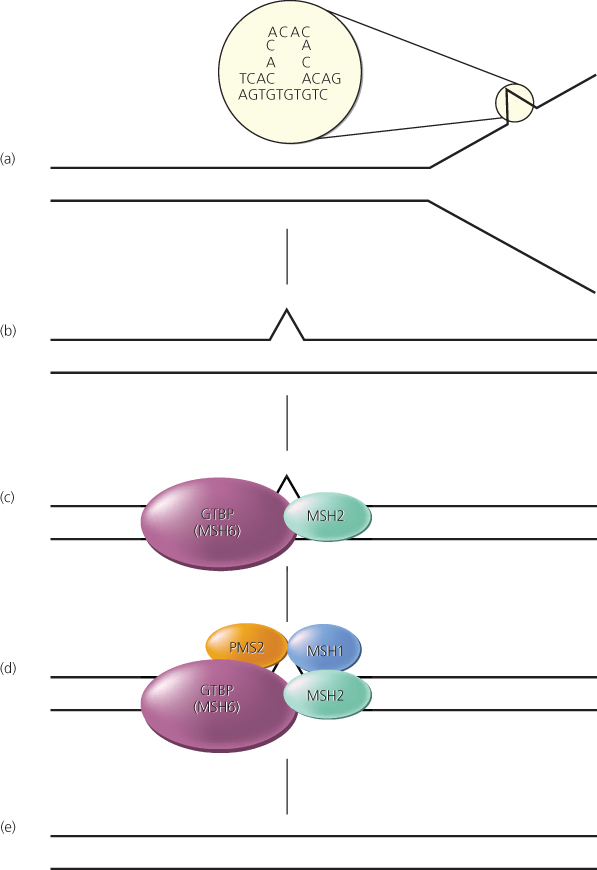
Figure 10 Mismatch repair pathway in human cells. (a,b) During DNA replication, DNA mismatches may arise, such as from strand slippage (shown) or misincorporation of bases (not shown). (c) The mismatch is recognized by MutS homologs, perhaps most often Msh2 and GTBP/Msh6, although another MutS homolog, Msh3, may substitute for GTBP/Msh6 in some cases. (d,e) MutL homologs, such as Mlh1 and Pms2, are recruited to the complex, and the mismatch is repaired through the action of a number of proteins, including an exonuclease, helicase, DNA polymerase, and ligase.
Germline mutations in the known MMR genes have been detected in only 2–4% of colorectal cancer patients, although approximately 15–20% of all colon cancers display the MSI phenotype.226, 229–234 Clearly, only a small fraction of the sporadic colorectal cancers with the MSI phenotype develop as the result of a germline mutation in a known MMR gene. Somatic mutations in MMR genes have been found in some sporadic colorectal cancers with the MSI phenotype.235 In most sporadic cases, however, inactivation of the MLH1 gene occurs as a result of epigenetic inactivation.236, 237
Many of the mutations arising in cells with MMR deficiency are likely to be detrimental to cell growth. A small fraction of the total mutations that arise presumably activate oncogenes or inactivate tumor suppressor genes. Some genes are preferentially mutated in MMR-deficient cancers, presumably because the mutations confer a selective growth advantage. For instance, genes that contain repetitive DNA sequences, such as microsatellite tracts, might be expected to be targets of mutation in these cancers, and data support this prediction.
BRCA1 and BRCA2 genes
As in the case of colorectal cancers, family history has long been hypothesized to be a major risk factor in breast cancer. The greatest risk is seen in those who have a history of breast cancer in multiple first-degree relatives. However, only in the late 1980s was evidence obtained that predisposition to breast cancer in some families could be attributed to a highly penetrant autosomal dominant allele. In 1990, Hall and colleagues reported the localization of one such breast cancer predisposition gene, BRCA1, on chromosome 17q21.238 Others found that germline BRCA1 mutations substantially increase the risk not only of breast cancer but also of ovarian cancer.239, 240 Intensive research efforts were focused on the region of chromosome 17q harboring BRCA1, and the gene was ultimately identified by positional cloning approaches in 1994.241, 242
Studies of germline BRCA1 mutations in breast cancer patients have yielded important results. In studies of families with four or more cases of breast or ovarian cancer (or both) diagnosed before age 60 years, germline BRCA1 mutations were identified in nearly 50% of families studied.243–245 In fact, germline BRCA1 mutations may account for cancer predisposition in roughly 75% of families who manifest both breast and ovarian cancer.243, 244 Many distinct germline BRCA1 mutations have been identified, although most of the mutations result in the synthesis of a truncated BRCA1 protein.243, 244 Although most germline BRCA1 mutations have been identified in only one or a few families, some mutations have been found recurrently. The 11 most common mutations account for about 45% of the total BRCA1 mutations observed.244, 245 In fact, the two most common mutations in BRCA1 (185delAG and 5382insC) account for approximately 10% of the total. Of note, the 185delAG frameshift mutation at codon 185 of BRCA1, involving a deletion of two bases (adenine and guanine), has been identified in more than 20 Jewish families with familial breast or ovarian cancer. Moreover, population surveys of Ashkenazi Jews, chosen without regard to a family history of cancer, indicate that approximately 1% carry the 185delAG mutation.244–246 Based on studies of families with germline BRCA1 mutations, the lifetime risks of breast cancer and ovarian cancer in those carrying an inactivating mutation are estimated to be 85% and 50%, respectively.243, 244, 247 Whether specific germline BRCA1 mutations confer a greater risk of breast and ovarian cancer than do other mutations remains uncertain.
As with most tumor suppressor genes and their associated familial cancer syndromes, germline mutations of BRCA1 lead to the presence of a mutant allele in every cell of the body. Cancers then arise through inactivation of the second wild-type allele by the mechanisms outlined in Figure 2. In the case of BRCA1 (and BRCA2), LOH of the remaining wild-type allele is usually responsible for the second “hit” leading to inactivation of the BRCA1 gene. In addition, LOH of the BRCA1 locus was found in approximately 50% of unselected breast cancers and 65–80% of unselected ovarian cancers.244, 248 Because most breast and ovarian cancers are not associated with a hereditary predisposition (i.e., they are sporadic), these studies of unselected cancers led investigators to hypothesize that BRCA1 would play an important role in the development of sporadic breast and ovarian cancers. However, despite these LOH data, sporadic breast and ovarian cancers rarely harbor mutations in the BRCA1 gene, demonstrating that at least one wild-type allele is still present in most of these sporadic cancers.244, 248 Somewhat surprisingly, this finding would suggest that BRCA1 does not have a role in the genesis of the more common, nonfamilial forms of breast and ovarian cancers. However, it is still possible that downstream effectors of the BRCA1 protein are altered in sporadic breast and ovarian cancers, suggesting that the pathway, rather than the gene, is an important contributor to the carcinogenic process of these cancers.249
Although germline mutations in the BRCA1 gene underlie cancer predisposition in roughly 40–50% of families with multiple breast cancer cases, another highly penetrant autosomal dominant susceptibility gene termed BRCA2 plays a critical role in a significant fraction of the families with multiple breast cancer cases lacking BRCA1 mutations. The BRCA2 gene was mapped to chromosome 13q12-13 in 1994250 and identified by positional cloning strategies in 1995.251 Although germline mutations in BRCA1 and BRCA2 appear to confer essentially similar lifetime risks of female breast cancer (i.e., ∼80%), the risk of ovarian cancer is reduced to approximately 10% in those with BRCA2 mutations compared with approximately 40–50% in those with BRCA1 mutations. The risk of male breast cancer is markedly elevated in BRCA2 mutation carriers, with a lifetime risk of approximately 7%, as opposed to a 1% lifetime risk of in BRCA1 mutation carriers.252 There also appears to be an elevated risk of pancreatic and several other cancers in both male and female BRCA2 mutation carriers.244 As is the case for BRCA1, LOH of the BRCA2 locus at 13q12, but not at the RB1 locus at 13q14, has been observed in some sporadic breast, pancreatic, head and neck, and other cancers, suggesting that BRCA2 may be a target for somatic mutations in cancer. However, as with BRCA1, detections of somatic BRCA2 mutations in sporadic cancers have been few,244 once again suggesting that perhaps the pathway, rather than the gene, is the target of genetic alteration in sporadic forms of these cancers.
The BRCA1 and BRCA2 genes each encode a large nuclear protein. The amino acid sequences of the two proteins have only short regions of similarity with one another or other well-characterized proteins. Although their lack of obvious functional motifs stymied initial attempts to define the cellular functions of BRCA1 and BRCA2, several lines of evidence indicate that both proteins interact directly or indirectly with homologs of yeast Rad51, a protein that functions in the repair of double-stranded DNA breaks.253–260 Moreover, the BRCA1, BRCA2, and Rad51 proteins all appear to be present in a stable multiprotein complex in the cell’s nucleus. Consequently, it has been suggested that BRCA1 and BRCA2 may function in the response to or repair of DNA damage, particularly double-strand DNA breaks.261 Other findings imply that BRCA1 and perhaps BRCA2 may have a role in regulating transcription.262 Although the DNA repair and transcription regulation functions may be distinct, it is entirely possible that the two functions are linked in a process sometimes called transcription-coupled DNA repair.255 There is precedent for this idea, in that some nucleotide excision repair genes causing xeroderma pigmentosum can also function in transcription,263 and repair of certain types of DNA damage is known to be coupled to transcription.264
Candidate tumor suppressor genes
The tumor suppressor genes discussed earlier and others summarized in Table 1 are distinguished by the fact that germline-inactivating mutations in the genes are associated with inherited cancer predisposition. The link between germline mutation and elevated cancer risk provides incontrovertible evidence of the gene’s role in tumorigenesis. Other findings, such as the demonstration in sporadic cancers of LOH of one tumor suppressor gene allele accompanied by somatic mutation of the remaining allele, offer evidence for a more widespread role for many of the inherited cancer genes. Although the tumor suppressor genes in Table 1 are definitively linked to inherited cancer syndromes, it is possible that germline mutations in other bona fide tumor suppressor genes may be associated with a more modest cancer risk, as has already been demonstrated for the I1307K mutation in APC. A complete listing of all documented tumor suppressor genes is beyond the scope of this chapter and is ever-evolving as new data come to light from detailed sequencing studies of human cancers. Based on the prevalence of inactivating mutations identified in such studies, it is estimated that there are approximately 100 tumor suppressor genes that play a role in human cancers of various types.64 The majority of these genes are only inactivated somatically and do not give rise to inherited cancer predisposition syndromes.
Table 1 Select tumor suppressor and stability genes associated with inherited cancer predisposition syndromes
| Genea | Syndrome | Hereditary pattern | Pathwayb | Major hereditary tumor typesc |
| APC | FAP | Dominant | APC | Colon, thyroid, stomach, intestine |
| AXIN2 | Attenuated polyposis | Dominant | APC | Colon |
| CDH1 (E-cadherin) | Familial gastric carcinoma | Dominant | APC | Stomach |
| GPC3 | Simpson–Golabi–Behmel syndrome | X-linked | APC | Embryonal |
| CYLD | Familial cylindromatosis | Dominant | APOP | Pilotrichomas |
| EXT1, EXT2 | Hereditary multiple exostoses | Dominant | GLI | Bone |
| PTCH | Gorlin | Dominant | GLI | Skin, medulloblastoma |
| SUFU | Medulloblastoma predisposition | Dominant | GLI | Skin, medulloblastoma |
| FH | Hereditary leiomyomatosis | Dominant | HIF1 | Leiomyomas |
| SDHB, SDHC, SDHD | Familial paraganglioma | Dominant | HIF1 | Paragangliomas, pheochromocytomas |
| VHL | von Hippel–Lindau | Dominant | HIF1 | Kidney |
| TP53 (p53) | Li–Fraumeni | Dominant | TP53 | Breast, sarcoma, adrenal, brain, and so on |
| WT1 | Familial Wilms tumor | Dominant | TP53 | Wilms |
| STK11 (LKB1) | Peutz–Jeghers | Dominant | PI3K | Intestinal, ovarian, pancreatic |
| PTEN | Cowden | Dominant | PI3K | Hamartoma, glioma, uterus |
| TSC1, TSC2 | Tuberous sclerosis | Dominant | PI3K | Hamartoma, kidney |
| CDKN2A (p16INK4A, p14ARF) | Familial malignant melanoma | Dominant | RB | Melanoma, pancreas |
| CDK4 | Familial malignant melanoma | Dominant | RB | Melanoma |
| RB1 | Hereditary retinoblastoma | Dominant | RB | Eye |
| NF1 | Neurofibromatosis | Dominant | RTK | Neurofibroma |
| BMPR1A | Juvenile polyposis | Dominant | SMAD | Gastrointestinal |
| MEN1 | Multiple endocrine neoplasia type I | Dominant | SMAD | Parathyroid, pituitary, islet cell, carcinoid |
| SMAD4 (DPC4) | Juvenile polyposis | Dominant | SMAD | Gastrointestinal |
| BHD | Birt–Hogg–Dube | Dominant | ? | Renal, hair follicle |
| HRPT2 | Hyperparathyroidism jaw-tumor | Dominant | ? | Parathyroid, jaw fibroma |
| NF2 | Neurofibromatosis | Dominant | ? | Meningioma, acoustic neuromas |
| MUTYH | Attenuated polyposis | Recessive | BER | Colon |
| ATM | Ataxia telangiectasia | Recessive | CIN | Leukemias, lymphomas, brain |
| BLM | Bloom | Recessive | CIN | Leukemias, lymphomas, skin |
| BRCA1, BRCA2 | Hereditary breast cancer | Dominant | CIN | Breast, ovary |
| FANCA, FANCC, FANCD2, FANCE, FANCF, FANCG | Fanconi anemia A, C, D2, E, F, and G | Recessive | CIN | Leukemias |
| NBS1 | Nijmegen breakage | Recessive | CIN | Lymphomas, brain |
| RECQL4 | Rothmund–Thomson | Recessive | CIN | Bone and skin |
| WRN | Werner | Recessive | CIN | Bone and brain |
| MSH2, MLH1, MSH6, PMS2 | HNPCC | Dominant | MMR | Colon, uterus |
| XPA, XPC; ERCC2, ERCC3, ERCC4, ERCC5; DDB2 | Xeroderma pigmentosum | Recessive | NER | Skin |
a Representative genes of all major pathways and hereditary cancer predisposition types are listed. Approved gene symbols are provided for each entry; alternative names are in parentheses.
b In many cases, the gene has been implicated in several pathways. The single pathway that is listed for each gene represents a “best guess” (when one can be made) and should not be regarded as conclusive.
c In most cases, the nonfamilial tumor spectrum caused by somatic mutations of the gene includes those occurring in familial cases but also additional tumor types. For example, mutations of TP53 and CDKN2A are found in many more tumor types than those to which Li–Fraumeni and familial malignant melanoma patients, respectively, are predisposed.
Abbreviations: APC, adenomatous polyposis coli; APOP, apoptotic pathway; BER, base excision repair; CIN, chromosomal instability; FAP, familial adenomatous polyposis; GLI, glioma-associated oncogene; HIF1, hypoxia-inducing factor 1; MMR, mismatch repair; NER, nucleotide excision repair; PI3K, phosphatidylinositol 3-kinase; RB, retinoblastoma; RTK, receptor tyrosine kinase pathway; SMAD, SMA- and MAD-related protein 4; wt, wild type.
Source: Adapted from Vogelstein and Kinzler 2004.131 Reprinted by permission from Macmillan Publishers Ltd.
Studies of RNA have revealed a large number of genes whose transcription is reduced in cancers. Some of these genes were initially termed tumor suppressors solely on the basis of this reduced expression. A small subset of these genes may, indeed, have critical roles in growth regulation, but expression data provide insufficient evidence to invoke causality. The altered expression of genes in cancers more often reflects the effect rather than the cause of the neoplastic process (i.e., the altered growth and differentiation properties of cancer cells and their abnormal microenvironment compared with normal cells in the tissue or organ from which the cancer arose). Moreover, many genes that play no role in cancer can dramatically alter cell growth when expressed exogenously at high, unphysiologic levels, rendering functional analyses of such candidates problematic. In the end, the functional evidence should be carefully weighed before a conclusion can be drawn that a gene has a causal role in tumorigenesis and that it should appropriately be designated a tumor suppressor gene. The only definitive way to identify a tumor suppressor gene at present is to document a highly statistically significant number of inactivating mutations in it in specific cancer types.64
Summary
There is now overwhelming evidence that mutations in tumor suppressor genes are major molecular determinants for most common human cancers. Thus far, dozens of confirmed tumor suppressor genes have been identified by molecular cloning techniques. In some cases, these genes are inactivated in the germline, and their inactivation predisposes to cancer. Far more frequently, these same tumor suppressor genes are inactivated by somatic mutations during tumor development.
Although much has been learned about tumor suppressor genes, a great deal of work remains. Advancements in our understanding of tumorigenesis will continue with the detailed characterization of their normal cellular functions and the mechanisms through which mutations of these genes subvert these normal functions. It is essential to point out that tumor suppressor genes, unlike oncogenes, are in general not directly targetable by drugs: all drugs inactivate their target proteins and tumor suppressor genes are already inactivated in cancers. Therefore, the best hope for exploiting tumor suppressor gene mutations in cancers is through targeting downstream pathways that are consequently upregulated. Understanding the pathways through which these genes act is therefore of paramount importance.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


