Tumor Immunology
Introduction
Immune responses against tumor cells occur, in large part, because of expression of cell-surface components on malignant cells that do not occur with their normal counterparts and that are recognized as antigenic molecules. Experimental evidence for this phenomenon came from studies in mice, which showed that when tumor cells were injected subcutaneously into syngeneic (major histocompatibility complex [MHC]-matched) mice, the cells formed nodules that grew for a few days and then regressed. When identical tumor cells were reinjected into the mice, they did not produce nodules or grow. These findings were interpreted to mean that the mice that rejected the tumor did so because they had generated an immune response to the tumor. Subsequently, tumor-specific transplantation antigens (TSTAs), or as they are more commonly called, tumor antigens, have been demonstrated for many tumors in a variety of animal species, including humans.
The major focus of this chapter concerns the role of the immune system in tumor-cell destruction. It is believed that throughout life, tumor cells are generated in normal individuals and then destroyed by normal immune effector mechanisms, without notice or consequence. Obviously, these immunological mechanisms are not always successful. The hope is that our growing knowledge of host defense mechanisms and the phenomenon of immunosurveillance will provide new insights into how we might prevent and better treat cancer. The two major goals of tumor immunology are (1) to elucidate the immunologic relationship between the host and the tumor, and (2) to utilize the immune response to tumors for the purpose of diagnosis, prophylaxis, and therapy. We discuss various approaches to meeting these goals in this chapter.
Tumor Antigens
Advances in immunologic and molecular biologic methodology have greatly facilitated the identification of tumor antigens capable of eliciting immune reactions. Before defining the different categories of tumor antigens, it is important to underscore the principal biologic mechanisms that may lead to the appearance of immunogenic tumor antigens including mutation, gene activation, and clonal amplification. Like normal immune responses to foreign antigens, the immunogenic potential of tumor antigens is manifested when their expression stimulates immune effector mechanisms. The antigenic prerequisites that apply to foreign immunogens also apply to tumor antigens. As discussed in Chapter 4, a substance must possess the following characteristics in order to be immunogenic: (a) foreignness, (b) high molecular weight, (c) chemical complexity, and (d) degradability with the ability to interact with host MHC antigens. Immunogenic tumor antigens fulfill these criteria and thus have the potential to induce immune responses. Nonimmunogenic tumor antigens, on the other hand, are in many cases self-antigens to which some degree of tolerance exists. This poses major barriers to both de novo and vaccine-induced tumor immunity. Efforts to overcome these barriers include the use of peptides derived from self-antigens expressed on tumor cells that are engineered to contain altered amino acids in order to increase the immunogenicity of the tumor antigen.
Some tumor antigens consist of structures that are unique to the cancerous cells and are not present on their normal counterparts. Other tumor antigens may represent structures that are common to both malignant and normal cells but are masked on the normal cells and become unmasked on malignant cells. Still other antigens on malignant cells represent structures that are present on fetal or embryonic cells but disappear from normal adult cells. These latter antigens are referred to as oncofetal antigens. Some tumor antigens expressed on tumor cells represent structures that are qualitatively not different from those found on normal cells, but that are overexpressed and thus present at significantly higher levels on the cancer cell. These are typically products of oncogenes. An example of the latter is the high level of human epidermal growth factor receptor (HER) expression in certain breast and ovarian cancers due to overexpression of the HER-2/neu-1 oncogene. Another example is the elevated ras oncogene products present on some human prostate cancer cells. In each of these different cases, the structural features of the tumor antigens arising by these various mechanisms are often similar from individual to individual. This similarity has sometimes translated to therapeutic advances in the treatment of cancer since commonly expressed tumor antigens can serve as targets for immune-based therapy (e.g., monoclonal antibodies against HER-2 to treat individuals with breast and ovarian cancer). Finally, oncogenic retroviruses (e.g., human T-cell leukemia virus) that can transform normal cells into cancer cells also induce tumor cell antigens that exhibit extensive structural similarity.
Normal genes that were previously silent may also be activated by carcinogens. It is generally assumed that unique tumor antigens on tumors induced by carcinogens are products of mutated genes with hot spots for mutations. There is little or no cross-reactivity between carcinogen-induced tumors. This absence of cross-reactivity is probably due to the random mutations induced by the chemical or physical carcinogens, leading to a large array of different antigens. For example, if the chemical carcinogen methylcholanthrene is applied in an identical manner to the skin of two genetically identical animals, or on two similar sites on the same individual, the cells of the developing tumors (sarcomas, in this case) will exhibit antigens unique to each tumor, with no immunologic cross-reactivity between the tumors. As with chemically induced tumors, there is little or no cross-reactivity among physically induced tumors, such as those induced by ultraviolet light or by X-irradiation.
Carcinogens can also be responsible for conversion of an otherwise nonimmunogenic molecule to an immunogenic antigen together with clonal amplification of cells expressing these molecules. The carcinogen-induced transformation event(s) that cause the emergence of such expanded clones most likely affect the genes that possess mutation-sensitive hot spots while sparing the genes responsible for other normal proteins. When these normal proteins are clonotypic (i.e., only expressed by single clones of cells), their expression is dramatically amplified, therefore making them immunogenic, assuming tolerance can be broken. As an example, the idiotypes of antigen-specific receptors expressed by B or T cells may not be on sufficiently numerous cells to elicit a response in the normal host but may serve as target antigens on lymphoid tumor cells bearing the same idiotype.
The following two sections provide overviews of the various categories and examples of tumor antigens and the immune effector mechanisms that play a role in preventing tumor cell development. Our expanding knowledge of these areas of tumor immunology continues to facilitate the development of clinically useful tumor-specific immonotherapies.
Categories of Tumor Antigens
Tumor antigens may be classified into several major categories (Table 20.1). The categories differ in both the factors that induce the malignancy and the immunochemical properties of the tumor antigens.
Table 20.1. Categories of Tumor Antigens
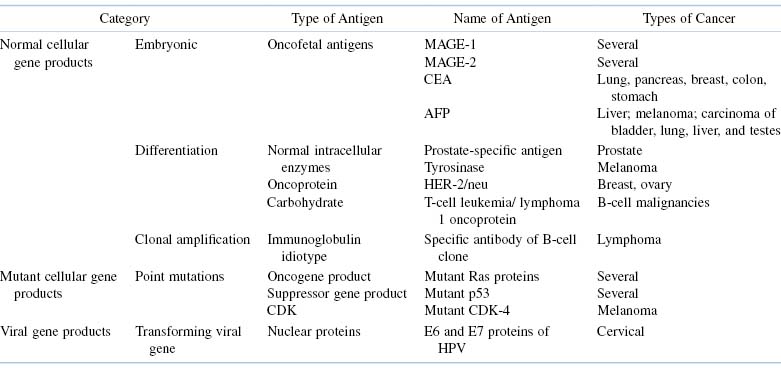
Normal Cellular Gene Products
Some tumor antigens are derived from normal genes that, under normal circumstances, are programmed to be expressed only during embryogenesis, namely, oncofetal antigens. Examples of these tumor antigens include the melanoma-associated antigen (MAGE) family of proteins that is not expressed in any normal adult tissues except for the testes (an immunologically privileged site). MAGE proteins are candidate tumor vaccine antigens because their expression is shared by many melanomas. Another category of oncofetal antigens is the group of cancer testes (CT) antigens. These are encoded by genes that are normally expressed only in the human germline but are also expressed in various tumor types, including melanoma, and carcinomas of the bladder, lung, and liver. Indeed, like many other tumor-associated antigens, the MHC restriction elements of the antigenic epitopes have been identified for both MAGE-1 and CT antigens. This information is being exploited in experiments aimed at developing immunogenic tumor peptide vaccines that can be presented by class I MHC antigens on antigen-presenting cells (APCs) to activate cytotoxic (CD8+) T cell responses.
Other examples of oncofetal antigens include the carcinoembryonic antigen (CEA) and α-fetoprotein (AFP). CEA is found primarily in serum of patients with cancers of the gastrointestinal tract, especially cancer of the colon. Elevated levels of CEA have also been detected in the circulation of patients with some types of lung cancer, pancreatic cancer, and some types of breast and stomach cancer. However, it should be noted that elevated levels of CEA have also been detected in the circulation of patients with nonneoplastic diseases, such as emphysema, ulcerative colitis, and pancreatitis, as well as in the sera of alcoholics and heavy smokers. AFP, which is normally present at high concentrations in fetal and maternal serum but absent from serum of normal individuals, is rapidly secreted by cells of a variety of cancers and is found particularly in patients with hepatomas and testicular teratocarcinomas.
Finally, amplified clones of malignant B or T cells that express antigen-specific receptors represent yet another example of how normal cellular gene products can be characterized as tumor antigens. The idiotype of the particular immunoglobulin or T-cell receptor (TCR) expressed by the transformed B or T cell, respectively, effectively identifies that clone as a unique population of malignant cells.
Mutant Cellular Gene Products
The genetic origins of several tumor antigens that are products of mutated genes have been identified. In every case, these antigens were the result of a somatic mutation (i.e., by a genetic change absent from autologous normal DNA). Often, these mutations occur in genes that encode functionally important parts of the expressed protein. There are several well-characterized examples of tumor antigens that are derived from mutant cellular gene products. Chronic myelogenous leukemia (CML) is characterized by the Philadelphia chromosome, a shortened chromosome 22 resulting from a reciprocal translocation between the bcr gene on chromosome 22 and the abl gene on chromosome 9 [t(9;22)]. The molecular equivalent of t(9;22) can be detected in virtually all cases of CML. It manifests with the expression of a bcr/abl fusion gene that encodes chimeric RNAs that produces copious amounts of the abl gene tyrosine kinase activity. This chimeric gene product, appears, at least in part, to be responsible for uncontrolled cell proliferation. In 2001, the U.S. Food and Drug Administration approved the use of the first Bcr-Abl tyrosine kinase inhibitor (TKI), namely, imatinib. Bcr-Abl tyrosine kinase TKIs are now the first-line therapy for most patients with CML.
Another example of a mutant cellular gene product is seen in many cases of familial melanoma. This disease is associated with a mutation in cyclin-dependent kinase-4 (CDK-4) that reduces binding to its inhibitor (p16INK-4), which happens to be a tumor-suppressor protein. Yet another example of a tumor antigen that is generated as a result of a mutant cellular gene is the mutant p53 protein. The p53 mutation generates common conformational changes in p53 protein that normally acts as a suppressor of cellular growth. Mutations in p53 are among the most common seen in tumors of human and experimental animals. They typically occur in evolutionarily conserved regions of the p53 gene and result in overexpression of the protein, which then serves as an immunogenic antigen for autologous B and T cells. Antibody and T-cell responses are also seen when mutations occur in ras oncogene-encoded proteins. Mutant ras proteins, resulting from a glycine substitution at position 12 of ras, represent one of the most common mutations in human cancers.
The potential use of mutated gene products as immunologic targets for immune-based therapies is best illustrated by experimental evidence showing that tumor immunity in vivo can be induced against normal p53 by mutant p53 peptides if administered to animals together with IL-12 to promote T-cell responses toward a TH1 phenotype. Because p53 is commonly overexpressed in cancer cells, the cytotoxic T-cell responses generated can destroy tumor cells. Furthermore, p53 knockout mice can be induced to generate cytotoxic T cells specific for normal p53 that, on adoptive transfer into p53 wild-type mice, can eradicate tumors overexpressing p53 without causing autoimmunity in the host.
Tumor Antigens Encoded by Oncogenes
Although a full discussion of carcinogenesis is beyond the scope of this chapter, it is important to summarize the oncogene theory in order to better understand the properties of certain oncogene-derived proteins that can be tumor antigens. All retroviral oncogenes are known to have close relatives in the genomes of virtually all normal vertebrate cells called c-onc genes or proto-oncogenes. The gene products of proto-oncogenes have been identified as proteins with known functions in normal cells, including growth factor receptors and signal transducers, to name a few. The oncogene theory postulates that when such proto-oncogenes are mutated or activated by other aberrant mechanisms, they overexpress or inappropriately express the mutated forms of their gene products, thereby contributing to neoplastic transformation and the development of cancer. Oncogenes are aberrantly activated in somatic cells in many forms of human cancer, including carcinoma and sarcomas, which include leukemias and lymphomas. The chief mechanisms of activation are chromosomal translocation, point mutation, and gene amplification. Table 20.2 gives a partial list of the known proto-oncogenes and their associated cancers.
Table 20.2. Activation of Cellular Proto-Oncogenes in Human Cancers

Animal studies have shown that tumors induced by oncogenic viruses exhibit extensive immunologic cross-reactivity. This is because any particular oncogenic virus induces the expression of the same antigens in a tumor, regardless of the tissue of origin or the animal species. For example, in animals, DNA viruses such as polyoma, SV40, and Shope papilloma virus induce tumors that exhibit extensive cross-reactivity within each virus group. Many leukemogenic viruses, such as Rauscher leukemia virus, induce the formation of tumors that exhibit cross-reactivity not only within each virus group but also between some groups. There is considerable evidence to suggest that several human cancers, such as Burkitt lymphoma, hepatocellular carcinoma, and nasopharyngeal carcinoma are caused by viruses. Adult T-cell leukemia is known to be caused by infection with human T-cell lymphotropic virus type 1 (HTLV-1).
As might be expected, the viral proteins, which ultimately serve as tumor antigens, are expressed intracellularly as predominantly nuclear proteins. In order for cytotoxic T lymphocytes (CTLs) to recognize these antigens, they must be processed and presented as class I MHC-associated peptides. Studies using SV40-specific CTL have confirmed that these cells can recognize processed fragments of proteins that are primarily located intracellularly. The unique tumor antigens of cells transformed by SV40 and several other viruses, including polyoma virus, adenovirus, and human papilloma virus (HPV) have been studied extensively and, in many cases, shown to be clearly related to the transformed phenotype and the establishment of malignancy. Such viruses have so-called early region genes designated E1A/E1B and E6/E7, which are transcribed during early stages of viral replication and in transformed cells by adenovirus and human papilloma virus, respectively. Like other categories of tumor antigens, these proteins are candidate targets for therapy.
Immunologic Factors Influencing the Incidence of Cancer
In the late 1950s, a hypothesis emerged to help explain the primary reason for development of T-cell–mediated immunity during the evolution of vertebrates. It was proposed that the main function of this arm of the immune system was to provide specific defense against altered self or neoplastic cells. The term immunosurveillance was coined to describe the concept of immunologic resistance against the development of cancer. However, there is growing recognition that immunosurveillance represents only one dimension of the complex relationship between the immune system and cancer. The concept of immunosurveillance is well supported by immunocompromised animal studies and epidemiologic studies of patients with various immunodeficiencies (primary, secondary, or acquired) that correlate an increased incidence of cancer but only with regard to cancers associated with viruses or, in some cases, UV exposure. By contrast, most common forms of cancer are not increased in immunocompromised individuals. However, patients with immunodeficiency diseases are usually susceptible to viral infections and certain malignant neoplasms (Table 20.3).
Table 20.3. Malignant Neoplasms with an Increased Incidence in Immunodeficiency Patients
| Type of Immunodeficiency | Cancer | Associated Virusa |
|---|---|---|
| Primary (congenital) | Hepatocellular carcinoma | HBV |
| B-cell lymphoma | EBV | |
| Secondary (e.g., drug-induced) | B-cell lymphoma | EBV |
| Squamous cell carcinoma (skin) | HPV | |
| Hepatocellular carcinoma | HBV | |
| Cervical carcinoma | HPV | |
| AIDS | Hepatocellular carcinoma | HBV |
| Cloagenic or oral carcinoma | HPV | |
| B-cell lymphoma | EBV |
aHBV, hepatitis B virus; EBV, Epstein–Barr virus; HPV, human papilloma virus
The absence of immunosurveillance of spontaneous cancers or those induced by carcinogens does not imply that such tumors do not express immunogenic tumor antigens. Indeed, there is sufficient evidence to support the conclusion that these tumor cells, like those induced by viruses, are sensitive to immunologic destruction. Nevertheless, the natural development of tumor-specific immune responses sometimes fails to prevent cancer from developing. Indeed, recent work has shown that the immune system may also promote the emergence of primary tumors with reduced immunogenicity that are capable of escaping immune recognition and destruction. This finding prompted the development of the cancer immunoediting hypothesis to more broadly encompass the potential host-protective and tumor-sculpting functions of the immune system throughout tumor development. Cancer immunoediting is a dynamic process composed of three phases: (1) elimination, (2) equilibrium, and (3) escape (Figure 20.1). In the first phase of elimination, cells and molecules of the innate and adaptive immune systems, which comprise the cancer immunosurveillance network, may eradicate the developing tumor and protect the host from tumor formation. However, if this process is not successful, the tumor cells may either enter the equilibrium phase where they may be maintained chronically or immunologically sculpted by immune “editors” to produce new populations of tumor-cell variants. These variants may eventually evade the immune system by a variety of mechanisms and become clinically detectable in the escape phase.
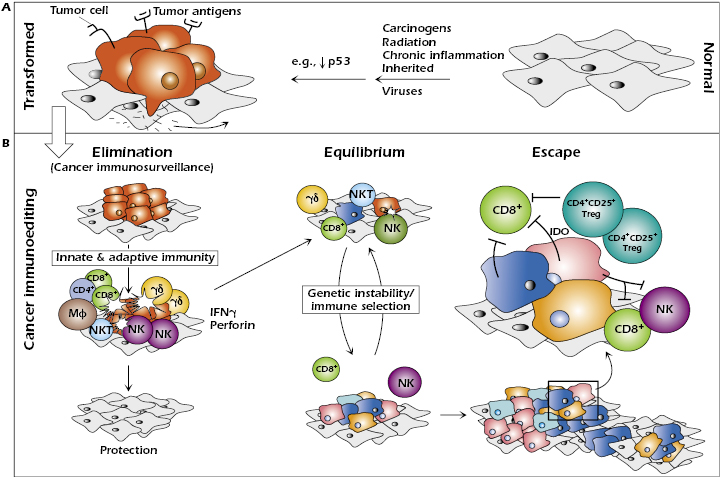
Elimination represents the classical concept of cancer immunosurveillance; equilibrium is the period of immune-mediated latency after incomplete tumor destruction in the elimination phase, and escape refers to the final outgrowth of tumors that have outstripped immunological restraints of the equilibrium phase.
Even at early stages of tumorigenesis, these cells may express distinct tumor-specific markers and generate proinflammatory “danger” signals that initiate the cancer immunoediting process (Figure 20.1). In the first phase of elimination, cells and molecules of innate and adaptive immunity, which comprise the cancer immunosurveillance network, may eradicate the developing tumor and protect the host from tumor formation. However, if this process is not successful, the tumor cells may enter the equilibrium phase where they may be either maintained chronically or immunologically sculpted by immune “editors” to produce new populations of tumor variants. These variants may eventually evade the immune system by a variety of mechanisms and become clinically detectable in the escape phase.
Effector Mechanisms in Tumor Immunity
Until recently, most of the information concerning tumor antigen-specific immune effector mechanisms and their capacity to destroy tumor cells has been derived from experiments with transplantable tumors in animals or from in vitro experiments. There is now ample evidence to suggest that adaptive and innate immune responses play important roles in the relationship between the host and the tumor in humans as well.
Immune effector mechanisms that are potentially capable of destroying tumors in vitro
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


