Tumor angiogenesis
John V. Heymach, MD, PhD  Amado Zurita-Saavedra, MD
Amado Zurita-Saavedra, MD  Scott Kopetz, MD, PhD
Scott Kopetz, MD, PhD  Tina Cascone, MD, PhD
Tina Cascone, MD, PhD  Monique Nilsson, PhD
Monique Nilsson, PhD
Overview
Angiogenesis, the growth of new capillary blood vessels, is central to cancer growth and metastasis and is recognized to be a potential therapeutic target for the treatment of cancer. Antiangiogenic agents have become part of the standard treatment armamentarium for many solid tumors, providing significant clinical benefits for some cancers (e.g., renal cell, colorectal) and modest or no benefit for others. This chapter is focused on principles of tumor angiogenesis that are intrinsic to the behavior of human cancer, and lessons that can be gleaned from the clinical testing and use of angiogenesis inhibitors to date.
Tumor angiogenesis
Angiogenesis, the growth of new capillary blood vessels, is central to the growth and metastatic spread of cancer. More than four decades ago, it was recognized to be a potential therapeutic target for the treatment of cancer.1 Since that seminal observation, the field has undergone explosive growth that has taken it from theory to clinical validation of angiogenesis as a therapeutic target, and antiangiogenic therapy is now in routine clinical use. Bevacizumab, a monoclonal antibody targeting vascular endothelial growth factor (VEGF), has undergone the most extensive clinical evaluation to date and is now a standard agent for the treatment of colorectal, lung, renal cell, and other malignancies. Several other antiangiogenic agents are also in routine use for other cancers including ramucirumab, an antibody targeting VEGF receptor-2; the VEGF- and placental-growth factor (PlGF) targeting protein aflibercept; and a number of tyrosine kinase inhibitors targeting angiogenic pathways including sunitinib, pazopanib, sorafenib, vandetanib, and axitinib.
While the progress of the field is encouraging, the clinical benefits of antiangiogenic agents have been relatively modest thus far, and key questions remain unanswered: How should antiangiogenic therapy be combined with other therapeutic regimens and treatment modalities? In what tumor types, and at what stage(s), should these agents be used? Can markers be developed to identify patients most likely to benefit, or experience toxicities, from treatment? The ultimate impact of antiangiogenic therapy in the treatment of cancer will be determined at least in part by the ability of basic researchers and clinicians to address these questions.
An understanding of the cellular and molecular basis of tumor angiogenesis is therefore important for clinicians who diagnose and treat cancer by whatever modalities. This chapter is focused on certain general principles of tumor angiogenesis that are intrinsic to the behavior of human cancer, and lessons that can be gleaned from the clinical testing and use of angiogenesis inhibitors to date.
Rationale for targeting tumor vasculature
The enormous progress that has been made in understanding molecular and genetic events that underlie the transformation of a normal cell to a cancer cell is reflected in many chapters in this book. Not surprisingly, most therapeutic approaches that have arisen from this research are targeted at the cancer cell, with a number of notable successes, such as therapies targeting BCR-ABL oncogene in CML or mutated c-KIT in gastrointestinal stromal tumors. In these cases it appears that cell survival and other key processes are highly dependent on a single activated pathway. For the vast majority of solid tumors, targeting a single molecular pathway in the cancer cell has produced much more modest benefits. Experimental and clinical evidence reviewed in this chapter indicates that it is prudent to develop cancer therapies against another target, the microvascular endothelial cell, with the understanding that the two targets are not mutually exclusive.
Consider a cancer cell that has progressed through a series of mutations so that by activation of certain oncogenes and by loss of specific suppressor genes, it has become self-sufficient in growth signals, insensitive to antigrowth signals, unresponsive to apoptotic signals, capable of limitless replicative potential, and tumorigenic.2 Current evidence argues that these neoplastic properties may be necessary but not sufficient for a cancer cell to expand into a population that is symptomatic, clinically detectable, metastatic, and lethal. For a tumor to develop a metastatic and/or a lethal phenotype, it must first recruit and sustain its own private blood supply, a process called tumor angiogenesis.2
A tumor can recruit vessels through at least four mechanisms: (1) co-option of existing vessels; (2) sprouting from existing vessels (angiogenesis); (3) formation of new vessels de novo, typically from bone marrow-derived cells in the adult (vasculogenesis); (4) intussusception, the insertion of interstitial tissue columns into the lumen of preexisting vessels (reviewed in Ref. 3).
Nonangiogenic tumors are harmless
There is an early stage of neoplastic development when tumors are not yet able to recruit new microvascular endothelial cells and cannot induce angiogenesis. As a result, most human tumors remain in situ and dormant at a microscopic size and are harmless to the host.4
Cancer without disease: prolonged survival with nonangiogenic, dormant tumors
Nonangiogenic tumors in humans stop expanding at a microscopic size of ∼1 mm or less. For over 100 years, pathologists performing autopsies on individuals who died of accidental causes have found that for a given age group, a large number of individuals harbor in situ carcinomas, while a very small percentage in that age group are diagnosed with cancer during life.5 For example, carcinoma in situ is found in the breasts of 39% of women aged 40–50 years who died of trauma, but only 1% are ever diagnosed with cancer in this age range. Carcinoma in situ of the prostate is diagnosed in 46% of men aged 60–70 years who died of trauma, but only 15% are diagnosed during life. One mechanism that keeps these in situ carcinomas in check may be host-derived factors that prevent switching to the angiogenic phenotype. Physiologic levels of endogenous angiogenesis inhibitors (see later sections) may serve this function.6
Disease of cancer requires expansion of tumor mass
Expansion of the tumor mass beyond the initial microscopic size of a nonangiogenic tumor, resulting in the disease of cancer, is usually dependent on recruitment of endothelial cells. Such endothelial cell recruitment begins when tumor cells undergo a switch to the angiogenic phenotype.7–9 The angiogenic phenotype is driven by a number of changes which may include (1) increased expression by tumor cells of angiogenic proteins such as VEGF and basic fibroblast growth factor (bFGF); (2) increased expression of angiogenic proteins from stromal cells (i.e., stromal fibroblasts), a process induced by the tumor itself; (3) decreased expression of endogenous angiogenesis inhibitors (i.e., thrombospondin [TSP]-1) by the tumor and by stromal fibroblasts; and (4) recruitment of bone marrow-derived endothelial cell precursors (see below for a detailed discussion of the switch to the angiogenic phenotype). Other mechanisms will unquestionably be uncovered. Absence of tumor angiogenesis prevents expansion of the tumor mass beyond a microscopic size, thereby avoiding metastatic spread and tumor-related symptoms, and hence, “cancer without disease”.6 The therapeutic implications of this concept are profound. Therapeutic blockade of angiogenesis may not only slow the growth of clinically evident tumors, but also may help prevent the emergence of microscopic lesions into clinically evident tumors.
Recruitment of microvascular endothelial cells is also necessary for expansion of a normal tissue mass and for expansion of an organ mass, for example, after partial hepatectomy.10 In fact, angiogenesis is fundamental to reproduction, development, and repair, but such physiologic angiogenesis occurs mainly as short-lived capillary blood vessel growth that usually lasts only days (ovulation angiogenesis), weeks (wound healing angiogenesis), or months (fetal and placental angiogenesis) but then it is always downregulated spontaneously and on a predictable timetable.11, 12 These physiological roles for angiogenesis help explain below some of the toxicities observed from use of angiogenesis inhibitors in the clinic.
Historic background
More than 100 years ago, it was observed that tumors were often more vascular during surgery than corresponding normal tissues.13 This was explained by simple dilation of existing host blood vessels.14 Vasodilation was thought to be a side effect of metabolites or of necrotic tumor products escaping from the tumor. Three reports, although largely overlooked, suggested that tumor hyperemia could be related to neovascularization and not solely to vasodilation.15, 16 Nonetheless, debate continued in the literature for two more decades about whether a tumor could expand to a large size (centimeters) by simply living on preexisting vasculature (vessel co-option).17 Even the few investigators who accepted the concept of tumor-induced neovascularization generally assumed that this was an inflammatory reaction, a side effect of tumor growth, not a requirement for tumor growth.18 It is now recognized that the two processes are often linked and that the recruitment of inflammatory cells plays a key role in initiating and promoting tumor angiogenesis.
Beginning of angiogenesis research
Hypothesis: tumor growth depends on angiogenesis
In 1971, Folkman proposed a new hypothesis that tumor growth is angiogenesis dependent.1 This hypothesis suggested that tumor cells and vascular endothelial cells within a neoplasm may constitute a highly integrated ecosystem and that endothelial cells may be switched from a resting state to a rapid growth phase by a “diffusible” chemical signal from tumor cells. An additional speculation was that angiogenesis could be a relevant target for tumor therapy (i.e., antiangiogenic therapy). These ideas were based on experiments Folkman and Frederick Becker performed in the early 1960s, which revealed that tumor growth in isolated perfused organs was severely restricted in the absence of tumor vascularization (Figure 1).19–23
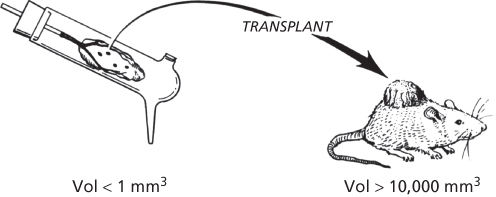
Figure 1 Tumors remain avascular in isolated perfused organs.20 Whole organs, supported by perfusion, allow growth of tumors in isolation from a host. The tumor remains very small, less than 1 mm3, in an avascular environment compared to growth in mice which can exceed 10,000 mm3.
These concepts were not widely accepted at the time. Another obstacle to research on tumor angiogenesis was the conventional wisdom at that time that any new vessels induced by a tumor, `such as new vessels in a wound, would become established and thus could not be made to involute. From this assumption, scientists concluded that antiangiogenic therapy could never regress a tumor; therefore, it would be fruitless to try to discover angiogenesis inhibitors. Eventual acceptance of Folkman’s 1971 hypothesis, and the development of angiogenesis research as a field, was subsequently facilitated by a number of advances including the ability to reproducibly culture vascular endothelial cells, the discovery of endogenous angiogenesis proteins, and the identification of drugs able to block angiogenesis in vitro and in vivo.24–28
Throughout the 1970s, laboratory studies were devoted to demonstrating that tumor vessels were new proliferating capillaries; the sequential steps of the angiogenic process could be identified; qualitative and quantitative bioassays for angiogenesis could be developed; viable tumor cells released diffusible angiogenic factors that stimulated new capillary growth and endothelial mitosis in vivo, despite the arrest of tumor cell proliferation by irradiation; necrotic tumor products were not angiogenic per se; and angiogenesis itself could be inhibited.29–32 Today the field has broadened to include a wide spectrum of basic science disciplines, from developmental biology to molecular genetics, as well as a variety of clinical specialties, including oncology, cardiology, dermatology, gynecology, ophthalmology, and rheumatology.
Experimental evidence
By the mid-1980s, considerable experimental evidence had been assembled to support the hypothesis that tumor growth is angiogenesis dependent. The idea could now be stated in its simplest terms: “Once tumor take has occurred, every further increase in tumor cell population must be preceded by an increase in new capillaries that converge upon the tumor”.33 The hypothesis predicted that if angiogenesis could be completely inhibited, tumors would become dormant at a small, possibly microscopic, size.22 It forecasted that while the presence of neovascularization would be necessary but not sufficient for expansion of a tumor, the absence of neovascularization would prevent expansion of a primary tumor mass beyond 1–2 mm3 and restrict a metastasis to a microscopic dormant lesion (Figure 1).
The hypothesis that tumors are angiogenesis dependent has been supported by a large body of preclinical evidence,34 including pharmacological and genetic studies, as well as clinical studies discussed in a later section. Some of the observations supporting the angiogenic hypothesis included:
- 1. Tumors implanted into subcutaneous transparent chambers grow slowly before vascularization, and tumor volume increases linearly. After vascularization, tumor growth is rapid and tumor volume may increase exponentially.35, 36
- 2. Tumors grown in the vitreous of the rabbit eye remain viable but are restricted to diameters of less than 0.50 mm for as long as 100 days. Once such a tumor reaches the retinal surface, it becomes neovascularized and within 2 weeks can undergo a 19,000-fold increase in volume over the avascular tumor.37
- 3. The limit of oxygen diffusion is approximately 100–200 µm. Tumor cells that exceed these distances from a capillary vessel become necrotic, as determined by intravital microscopy of tumors in transparent skin chambers in mice (Figure 2).38
- 4. In transgenic mice that develop carcinomas of the β cells in the pancreatic islets, large tumors arise from a subset of preneoplastic hyperplastic islets but only after they have become vascularized.8
- 5. In colon carcinomas arising in rats after carcinogen exposure, there is an early phase (tumor diameter < 3.5 mm) during which the tumor is temporarily supplied by preexisting host microvessels that are dilated and widened.39 This stage is similar to “co-option” of blood vessels.40 Subsequently, new capillary vessels sprout and proliferate (angiogenesis), which leads to increasing microvessel density and is accompanied by rapid tumor growth.
- 6. A neutralizing antibody to VEGF was administered to mice bearing tumors that induced angiogenesis solely by VEGF.41 Tumor growth was inhibited by more than 90%. The antibody had no effect on the tumor cells in vitro.42 This observation has been replicated using a variety of different means to block VEGF, including a fusion protein engineered from VEGF receptors (VEGF trap).43 Similar results were seen with an antibody directed against another angiogenic factor, bFGF.44
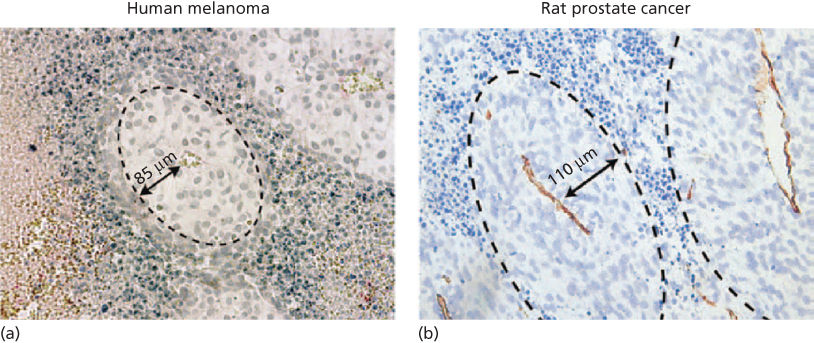
Figure 2 (a) A cuff of viable tumor cells surrounding a microvessel in a human melanoma growing in a SCID mouse has an average radius of 85 µm. The appearance of an ellipsoid is a result of the way the section is cut. (b) A cuff of rat prostate cancer cells surrounding a microvessel has an average radius of 110 µm.
Biology of tumor angiogenesis
The role of angiogenesis in preneoplasia and early tumorigenesis
In the experiments with isolated perfused organs by Folkman and colleagues more than four decades ago, the growth of tumors was severely restricted in diameter in the absence of angiogenesis.20, 45 This and other studies led to the proposal that the growth of solid tumors is dependent on new capillary sprouts (termed angiogenesis), and that without angiogenesis solid tumors might become completely dormant.1 This raised the possibility that angiogenesis might be a therapeutic target not only for the treatment of advanced cancers, but potentially for chemoprevention as well. In recent years, angiogenesis has been validated as a therapeutic target for advanced cancers through a growing body of preclinical and clinical studies. The study of angiogenesis inhibitors for blocking the earliest steps in the development of cancer (chemoprevention) has lagged behind, despite the strong support for the concept provided by preclinical studies (reviewed in Refs 46 and 47).
Preclinical studies demonstrating a role for angiogenesis in early tumorigenesis
In the 1970s it was shown by Gullino48 that precancerous tissues demonstrate signs of early angiogenesis, and it was suggested that blocking this process may be used to prevent cancer. Later, using transgenic mouse models of tumorigenesis, Hanahan and colleagues demonstrated that some premalignant lesions undergo “angiogenic switch” early in carcinogenesis, reflecting a change in the net balance between angiogenic stimulators and inhibitors.8, 9 VEGF and matrix metalloproteinase-9 (MMP-9) were shown to play important roles in the angiogenic switch in these models.49–51 In a murine model of pancreatic islet carcinoma, four different antiangiogenic agents were tested (TNP-470, endostatin, angiostatin, or the MMP inhibitor BB-94) and had distinct activity profiles in terms of their ability to prevent tumor formation (chemoprevention), slow the growth of small tumors (early intervention), and regress established tumors.8, 52, 53 In another study, established mammary cancer cells (i.e., previously transformed cells but not primary tumors) were implanted in a dorsal skinfold window chamber in rats, and were found to induce angiogenesis long before the tumor population would have reached the limiting size of a nonneovascularized tumor, that is, 0.2–2 mm. These and other preclinical studies suggest that angiogenesis is an early and critical step in tumorigenesis, and that antiangiogenic agents may inhibit tumor progression and growth.
Preneoplasia is associated with increased angiogenesis in human tumors
Studies from human clinical specimens provide further support for the hypothesis that angiogenesis occurs early in tumor progression, typically during the preneoplastic stage. In cervix cancer, an initial mild increase in vessel density has been detected in the early dysplastic [cervical intraepithelial neoplasia (CIN) I] stage. Mid–late dysplasias (CIN II–III) exhibited a readily apparent angiogenic switch, wherein new vessels became densely apposed along the basement membrane underlying the dysplastic epithelium.54, 55 Biopsies from lung cancer patients and high risk individuals have shown that preneoplastic lesions ranging from hyperplasia and metaplasia to carcinoma in situ are associated with increased microvessel density in the surrounding mucosa.56–58 A distinctive pattern known as angiogenic squamous dysplasia57 has also been identified. The specific angiogenic stimulators in bronchial preneoplastic lesions have not been established but elevated levels of VEGF,56 EGFR (epidermal growth factor receptor),59 and COX-260 have been observed.
Role of angiogenesis in the metastatic spread of cancer
In addition to its role in enabling the growth of small premalignant or malignant lesions, angiogenesis contributes to the hematogenous metastatic spread of tumors. This spread may be facilitated by the presence of “mosaic” blood vessels in tumors, in which both endothelial and tumor cells form the luminal blood surface, facilitating the shedding of tumor cells into the circulation. In one study, approximately 15% of vessels in a colon cancer xenograft model were mosaic vessels in which tumor cells appeared to directly contact the luminal vessel surface without endothelial cells acting as a barrier.61 Similar numbers of mosaic tumor vessels were detected in human tumor biopsies. These observations suggest that the irregular architecture and function of tumor vessels may facilitate tumor cell shedding into the circulation and metastatic spread.
Micrometastases also appear to be dependent on angiogenesis in order to progress into clinically evident tumors. They may remain dormant in distant sites for an extended period of time but a small fraction of these acquire an adequate blood supply to permit the development of tumors; angiogenesis inhibitors can inhibit this process in a variety of murine models.62–67 The continued growth of both the primary tumor and metastases thereafter depends on the maintenance of an adequate blood supply. In theory, angiogenesis inhibitors may therefore offer benefit when used for chemoprevention as well as in treatment of early stage, occult metastatic, or advanced disease.46, 68, 69
Taken together, these preclinical and clinical studies provide evidence that the induction of angiogenesis is an early and important step in tumor progression, likely occurring in precancerous lesions, and is involved in metastatic spread. For these reasons, angiogenesis is a rational target for chemoprevention. Additional studies will be needed to elucidate the key regulators of early angiogenesis, and to identify the different types of antiangiogenic agents that may be optimal for chemoprevention, advanced disease, and other applications.
Regulators of angiogenesis
The discovery of diffusible factors stimulating tumor angiogenesis
The observation in the 1970s that tumors implanted into the avascular cornea or onto the vascularized chick chorioallantoic membrane induced an ingrowth of new capillaries indicated that tumors released diffusible angiogenic factors.9 This result motivated the development of in vitro and in vivo bioassays to guide the search for tumor-derived angiogenic factors.70
Fibroblast growth factors
bFGF (or FGF-2) was the first angiogenic protein to be isolated and purified from a tumor (1982), followed shortly by acidic FGF (aFGF or FGF-1).28, 71, 72 Acidic and bFGFs stimulate endothelial cell mitosis and migration in vitro and are among the most potent angiogenic proteins in vivo. They have high affinity for heparin and heparan sulfate.73 The expression of bFGF receptors is very low. A wide variety of cancer types secrete bFGF, including those of the central nervous system, sarcomas, genitourinary tumors, and even endothelial cells in the tumor vasculature. Identification of an FGF-binding protein secreted by tumors into the extracellular matrix may illuminate a mechanism of tumor mobilization of stored FGFs.74–76 Furthermore, some tumors recruit macrophages77 and activate them to secrete bFGF,78 whereas others attract mast cells, which, because of their high content of heparin, could sequester bFGF. In spontaneous tumors that arise in transgenic mice, aFGF and bFGF are exported into conditioned medium by angiogenic tumor cells but not by preangiogenic cells in earlier stages of tumor progression.38, 79 bFGF also interferes with adhesion of leukocytes to endothelium, and it has been suggested that tumors that elaborate bFGF may produce a form of local immunologic tolerance.80–82 In mouse models of cancer, inhibition of the FGFR signaling pathways results in inhibited tumor angiogenesis and reduced tumor growth.83
Abnormally elevated levels of bFGF are found in the serum and urine of cancer patients and in the cerebrospinal fluid of patients with different types of brain tumors.84, 85 High bFGF levels in renal carcinoma correlated with a poor outcome.86 Moreover, bFGF levels in the urine of children with Wilms’ tumor correlate with stage of disease and tumor grade.87
In addition to contributing to tumor angiogenesis, multiple FGFR fusion proteins have been identified on diverse cancers including lung squamous cell, bladder, glioblastoma, and thyroid cancer, and appear to be oncogenic drivers as pharmacologic inhibition of FGFR suppresses the growth of FGFR fusion-positive tumor models.88 Additional reports have shown FGFR1 to be amplified in a subset of small cell lung cancers among other malignancies, and tumor cells harboring FGFR amplification are sensitive to target inhibition.89 In these settings, inhibition of FGFRs will therefore have both direct antitumor cell effects and indirect tumor cell effects through targeting of the tumor vasculature.
Vascular endothelial growth factor (VEGF) family
Dvorak first proposed that tumor angiogenesis is associated with increased microvascular permeability.90 This led to the identification of vascular permeability factor (VPF).91–93 VPF was subsequently sequenced by Ferrara and colleagues and in 1989 was reported to be a specific inducer of angiogenesis called VEGF.92–94 At the same time, a novel angiogenic protein was first isolated and purified from a tumor (sarcoma 180) in the Folkman lab, and in a collaboration with Ferrara was shown to be VEGF.94 Since then, more than 40 angiogenic inducers have been identified, most as tumor products (Table 1).3, 95 VEGF is an endothelial cell mitogen and motogen that is angiogenic in vivo.96–98 Its expression correlates with blood vessel growth during embryogenesis and is essential for development of the embryonic vascular system.99, 100 VEGF expression also correlates with angiogenesis in the female reproductive tract, and in tumors.49, 101–103 VEGF is a 40–45 kDa homodimeric protein with a signal sequence secreted by a wide variety of cells and the majority of tumor cells. VEGF exists as five different isoforms of 121, 145, 165, 189, and 206 amino acids, of which VEGF165 is the predominant molecular species produced by a variety of normal and neoplastic cells. Two receptors for VEGF are found mainly on vascular endothelial cells, the 180 kDa fms-like tyrosine kinase (Flt-1)104 and the 200 kDa human kinase insert domain-containing receptor (KDR) and its mouse homolog, Flk-1.105 VEGF binds to both receptors, but KDR/Flk-1 transduces the signals for endothelial proliferation and chemotaxis.106–109 Other structural homologs of the VEGF family include VEGF-B, VEGF-C, VEGF-D, and VEGF-E.110, 111 VEGF-C and VEGF-D bind to Flt-4, which is preferentially expressed on lymphatic endothelium.112, 113
Table 1 Examples of regulators of angiogenesis
| Proangiogenic molecules | Antiangiogenic molecules | Transcription factors, oncogenes, and other regulators |
| Vascular endothelial growth factor (VEGF) | Interferon−α,β,γ | Hypoxia-inducible factor (HIF)-1α, 2α |
| Basic fibroblast growth factor (bFGF) | Thrombospondin-1,2 | Nuclear factor-κB (NFκB) |
| Transforming growth factor-α (TGF-α) | Angiopoietin 2 | Epidermal growth factor receptor (EGFR) |
| Platelet-derived growth factor (PDGF) | Tissue inhibitors of MMPs (TIMPs) | Ras |
| Epidermal growth factor (EGF) | Endostatin | p53 |
| Angiopoietins | Angiostatin | von Hippel-Lindau |
| Interleukin-6 | Interleukin-12 | Cadherin |
| Interleukin-8 | Endostatin | Integrin |
| Matrix metalloproteinases (MMPs) | Thrombospondin-1 | Semaphorin |
| Hepatocyte growth factor (HGF) | Id1, Id2 | |
| Stromal cell-derived factor-1α (SDF-1α) | Prolyl hydroxylases | |
| Delta-like ligand 4 (DLL4) | myc | |
| Ephrins | ||
| Monocyte chemoattractant protein-1 and other chemokines |
Klagsbrun et al. discovered that neuropilin-1, a neuronal guidance molecule, is a coreceptor for VEGF165, but not for VEGF.114, 115 This finding provides a molecular mediator that coordinates growth in the vascular system with the nervous system. Other neuronal guidance proteins and/or their receptors are also angiogenesis regulators. For example, neuropilin is a receptor for VEGF and for semaphorin. Semaphorin repels neurite outgrowth and is also an angiogenesis inhibitor. Ephrins are neural guidance molecules, but also genetically determine arteries and veins during embryogenesis. EphrinB2 is expressed by tumor vascular endothelium. Perhaps in part because of the overlap between regulators of the nervous and vascular systems, angiogenesis in brain tumors has distinctive characteristics such as high interstitial fluid pressure (IFP) and low oxygen tension that contribute to the malignant behavior of these tumors.116
Neuropilin is not a tyrosine kinase receptor and is expressed on nonendothelial cells, including tumor cells. This allows VEGF that is synthesized by tumor cells to bind to their surface. Surface-bound VEGF could make endothelial cells chemotactic to tumor cells, or it could act in a juxtacrine manner to mediate co-option of microvessels by tumor cells. Neuropilin also binds placenta growth factor-2 (PlGF-2), and heparin is essential for the binding of VEGF165 and PlGF-2 to neuropilin-1.40, 117 The natural cell surface polysaccharide in vivo is heparan sulfate, not heparin. Heparan sulfate may act as a template to accelerate the interaction of VEGF or PlGF-2 with VEGF. VEGF expression by tumors is upregulated by hypoxia and is often elevated near areas of tumor necrosis.118–121 Hypoxia activates hypoxia-inducible factor-1 (HIF-1), which binds to the hypoxia response element (HRE sequence) in the VEGF promoter, which leads to VEGF mRNA transcription.106, 120 Independent of this, hypoxia stabilizes the VEGF mRNA.
VEGF signal transduction
While VEGF-A binds VEGFR1 with a higher affinity than VEGFR-2, the majority of biological effects of VEGF-A on tumor endothelium are thought to be mediated through VEGFR-2. Upon ligand binding, VEGFR-2 dimerizes, resulting in activation of the tyrosine kinase and autophosphorylation of residues including Tyr951, Tyr996, Tyr1054, Tyr1175, and Tyr1214.122 Phosphorylation of these residues induces the activation of signal transduction molecules including PI3K, phospholipase C-γ (PLC-γ), Akt, Src, Ras, and MAPK (Figure 3). Phosphorylation of Tyr1175 results in the binding and phosphorylation of PLC-γ, which subsequently promotes the release of Ca2+ from internal stores and activation of protein kinase C (PKC). PKC activation and Ca2+ mobilization are considered to be critical for VEGF-A-induced cell proliferation and nitric oxide production, respectively.123
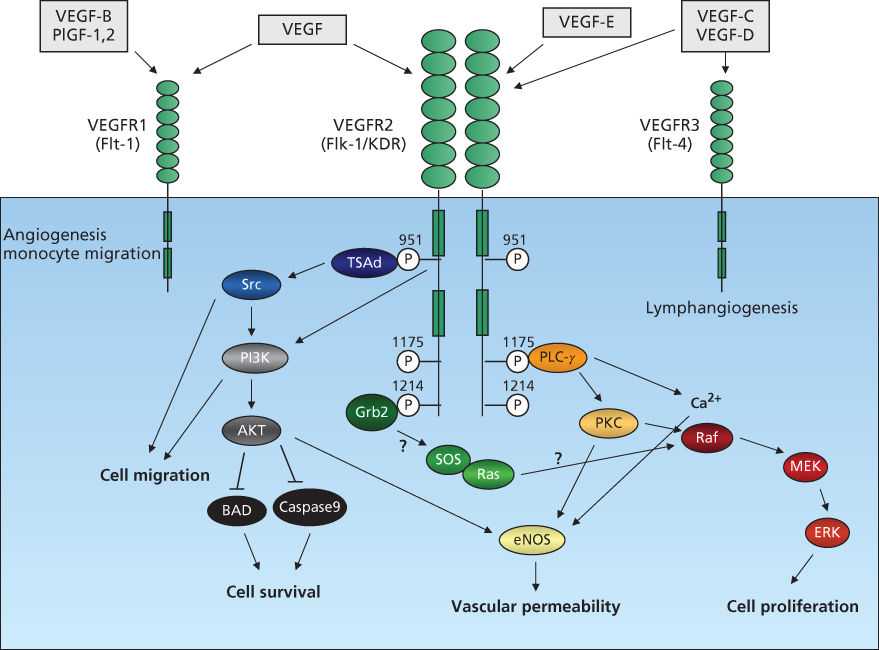
Figure 3 VEGFR signal transduction. VEGF family members, VEGF, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and PlGF bind three VEGFR tyrosine kinases, resulting in dimerization, receptor autophosphorylation, and activation of downstream pathways. Signal transduction via VEGFR2 is shown. Ligand binding to VEGFR2 activates signal-transduction molecules phospholipase C-[γ] (PLC-[γ]), PI3K, Akt, Ras, Src, and MAPK and regulates cell proliferation, migration, survival, and vascular permeability.
The PI3K pathway is paramount in the regulation of cell proliferation, survival, and migration. VEGF-A has been shown to promote phosphorylation of the p85 subunit of PI3K and enhance PI3K enzymatic activity. The mechanism by which VEGF-A results in activation of PI3K remains unclear, although studies have implicated a role for Src kinases, β-catenin, and VE-cadherin.124, 125 VEGFR-2-induced activation of PI3K results in accumulation of phosphatidylinositol-3,4,5-trisphosphate (PIP3), which induces phosphorylation of Akt/PKB. Once activated, Akt/PKB phosphorylates and thus inhibits proapoptotic proteins BAD and caspase-9.
Members of the Src family kinases, Src, Fyn, and Yes, are expressed in endothelial cells. Following VEGFR-2 autophosphorylation, T cell-specific adapter (TSAd) binds Tyr951 and then associates with Src. Src kinases control actin stress fiber organization and may mediate VEGF-A-induced PI3K activation. Ligand binding to VEGFR-2 also initiates activation of the Ras pathway, triggering signaling through the Raf-1–MEK–ERK signal cascade122 known to be important in growth factor-induced cell proliferation. This activation may occur through multiple routes.122, 126
Biological function of VEGF
In its initial discovery, VEGF was identified as a mediator of vessel permeability.110, 127 This capacity to render small veins and venules hyperpermeable is a critical function of VEGF. VEGF is indeed one of the most potent regulators of vascular permeability, and the observation that tumor-associated blood vessels are hyperpermeable is attributed to tumor-secreted VEGF. While the mechanisms by which VEGF increases microvascular permeability are incompletely understood, it may be at least in part due to VEGF-induced endothelial fenestrations,100 opening of junctions between adjacent endothelial cells,128 and through a calcium-dependent pathway involving nitric oxide (NO) production.129–131
In addition to its effect on vascular permeability, VEGF is a survival factor for endothelial cells, inhibiting endothelial cell apoptosis through activation of the PI3K–Akt pathway132 and increases in the antiapoptotic protein bcl-2.133 In vivo, VEGF blockade has been demonstrated to cause increases in apoptosis of immature, nonpericyte-covered vessels.134 VEGF is an endothelial cell mitogen though VEGFR-2 mediates signal transduction through Erk1/2, JNK/SAPK, and possibly PKC.133, 135 While VEGF is not as potent an endothelial cell mitogen as other factors such as bFGF, it has a broader range of activity for processes critical for angiogenesis. VEGF induces expression of MMPs and serine proteinases involved in degradation of the basement membrane, which is necessary for endothelial cell sprouting and invasion.133 In addition, VEGF facilitates endothelial cell migration through FAK and p38 MAPK-induced actin reorganization.3, 128, 133, 136
Circulating VEGF may be one of the angiogenic signals by which tumors recruit bone marrow-derived cells, including endothelial progenitors and myeloid cells whose recruitment is thought to be mediated by VEGFR-2 and VEGFR-1, respectively.137–141 A growing body of evidence suggests that VEGFR-bearing bone marrow-derived cells contribute to initiating tumor formation, by creating a “metastatic niche”, and/or help promote tumor angiogenesis142–146 although there appear to be VEGFR-1-independent mechanisms as well.147 Circulating endothelial and myeloid cells are further being studied as potential biomarkers, as noted later in the chapter, and may contribute to resistance to VEGF pathway inhibitors.148, 149 It is worth noting that not all VEGF in the circulation may be tumor derived. VEGF serum concentrations closely correlate with platelet counts in cancer patients. VEGF is stored in platelets, and is transported by and released from them.150 Furthermore, Pinedo and colleagues report that higher platelet counts correlate with a worse prognosis for cancer patients.151, 152 Therefore, it is possible that for those types of tumors that recruit bone marrow-derived endothelial cells, communication from tumor to bone marrow may be mediated in part by the VEGF in circulating platelets.
Angiopoietins
Angiopoietin-1 (Ang1) is a 70 kDa ligand that binds to a specific tyrosine kinase expressed only on endothelial cells, called Tie2 (also called Tek). Ang1 ligand binds Tie2 expressed on ECs, and Tie2 subsequently dimerizes and is phosphorylated. Activated Tie2 triggers activation of PI3K and Ras/Raf/MEK, promoting EC survival and proliferation/migration (Figure 4).153 A ligand for Tie1 has not been elucidated.114, 154–157 Like VEGF, Ang1 is an endothelial cell-specific growth factor. Ang1, however, is not a direct endothelial mitogen in vitro. Rather, it induces endothelial cells to recruit pericytes and smooth muscle cells to become incorporated in the vessel wall. Pericyte and smooth muscle recruitment is mediated by endothelial production of PDGF-BB (and probably other factors) when Tie2 is activated by Ang1.158 There is increased vascularization in mice that overexpress Ang1 in the skin.157 The vessels are significantly larger than normal and the skin is reddened. The vessels are not leaky and there is no skin edema, in contrast to dermal vessels of mice overexpressing VEGF. In double transgenic mice expressing both Ang1 and VEGF in the skin, dermal angiogenesis is increased in an additive manner, but the vessels do not leak.159 This model closely approximates angiogenesis in healing wounds (i.e., relatively nonleaky vessels with pericytes and some perivascular smooth-muscle cells contained in the vascular wall). In contrast, tumor vessels are leaky and thin walled with a paucity of pericytes. Angiopoietin-2, produced by vascular endothelium in a tumor bed, blocks the Tie-2 receptor and acts to repel pericytes and smooth muscle.154 Nevertheless, tumor vessels remain thin “endothelium-lined tubes” even though some of these microvessels reach the diameter of venules. High levels of Ang2 are associated with metastases in melanoma patients, and in preclinical models, Ang2 promotes metastatic formation.160, 161 A key point is that angiopoietins and VEGF together play a role in angiogenesis, and the activity of both is context dependent with different activities observed in angiogenic versus mature vessels. Because VEGF plays a key role in the proangiogenic activity of Ang2, using combination inhibitors targeting both the Ang/Tie2 and VEGF receptor pathways may provide clinical benefit, a concept that has begun to be explored. However, given that angiopoietins can apply either pro- or antitumorigenic effects, depending on the context, the best targeting approach remains unclear.153
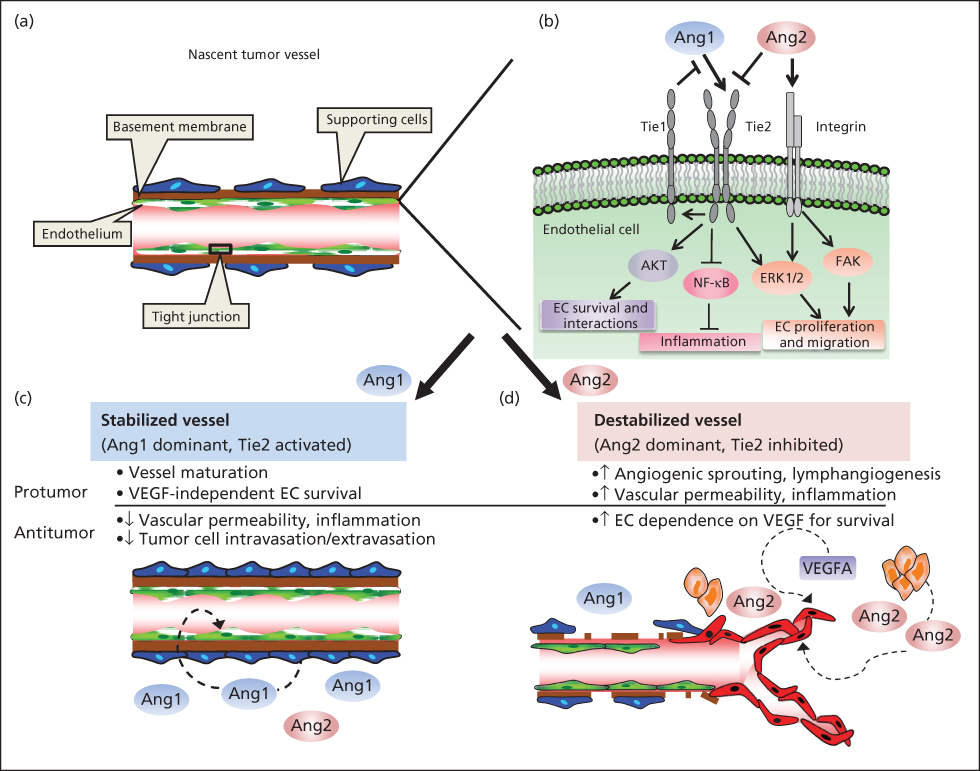
Figure 4 Ang/Tie2 signaling pathway. (a) Representation of nascent tumor blood vessel structure in quiescent state. Endothelial cells (ECs; green) are organized to form vessel lumen and tightly interact with perivascular cells (blue), separated by basement membrane. (b) Ang1 ligand binds Tie2 localized on ECs; Tie2 subsequently dimerizes and is phosphorylated. Activated Tie2 activates phosphatidylinositol 3-kinase and Ras/Raf/MEK, promoting EC survival and proliferation/migration. Generally, Ang2 functions as an Ang1 antagonist by inducing increased vascular permeability, sprouting vascularization. Autophosphorylated Tie2 activates Tie1, the extracellular domain of which in turn interferes with Ang1/Tie2 binding, therefore antagonizing Ang1 signaling. Ang1 has anti-inflammatory properties, whereas Ang2 plays a proinflammatory role. Ang1-activated Tie2 inhibits nuclear factor-κB–mediated inflammatory gene expression, whereas Ang2 promotes expression through blockade of Ang1 function. (c) Perivascular cells secrete Ang1, which binds Tie2 expressed on ECs. In this interaction, Ang1 contributes to vessel integrity, reduces vessel permeability, and maintains vasculature quiescence. (d) Ang2 plays a major role in angiogenesis by mediating dissociation of pericytes and destabilizing cellular junctions. In the presence of other proangiogenic factors (e.g., VEGF), ECs proliferate and/or migrate to form new sprouting, disorganized blood vessels. In the absence of proangiogenic factors, Ang2 signaling results in EC apoptosis and regression.
Source: Cascone and Heymach 2012.153 Reprinted with permission. © 2012 American Society of Clinical Oncology. All rights reserved.
Other factors regulating angiogenesis
Interleukin-8 (IL-8) is a proinflammatory chemotactic cytokine produced by monocytes, macrophages, and tumor cells.162 IL-8 induces endothelial cell proliferation and chemotaxis as well as promotes endothelial cell survival.162, 163 The effects of IL-8 are mediated through interactions with two cell-surface G protein-coupled receptors, CXCR1 and CXCR2, and activation of subsequent downstream signaling molecules including PI3K and MAPK.164 Hepatocyte growth factor/scatter factor (HGF/SF) is the ligand for c-Met.165 Originally identified as a mitogen for primary cultured hepatocytes, HGF has since been shown to facilitate tumor angiogenesis. c-Met is expressed on endothelial cells, and activation by tumor-secreted HGF/SF augments matrix degradation and endothelial cell invasion. NOTCH proteins and ligands are also elevated in several cancers, and NOTCH signaling has been shown to both promote and suppress tumor growth depending on the tumor type.166 Interactions between NOTCH and its ligand DLL4 promote tumor angiogenesis through an intricate cross-regulation of NOTCH and VEGF/VEGFR signal transduction pathways.167, 168
Endogenous inhibitors of angiogenesis
Endogenous angiogenesis inhibitors block vascular endothelial cells from proliferating, migrating, or increasing their survival in response to a spectrum of proangiogenic proteins, including VEGF, bFGF, interleukin-8, PDGF, and PD-ECGF. Natural angiogenesis inhibitors include interferon-α, interleukin-12, platelet factor 4, TSP-1, angiostatin, endostatin, arrestin, canstatin, tumstatin, PEX (MMP-2), pigment epithelium-derived factor, and antiangiogenic antithrombin III (an internal fragment of antithrombin III, named aaAT).27, 169–175
The first clue to the existence of endogenous angiogenesis inhibitors came from the discovery that interferon-α inhibited endothelial cell migration and that platelet factor 4 inhibited endothelial proliferation.176–178 Both were subsequently shown to inhibit angiogenesis.176–180 However, Bouck and her colleagues were the first to show that a tumor could generate an angiogenesis inhibitor; they subsequently proposed that the angiogenic phenotype was the result of a net balance of endogenous inhibitors and stimulators of angiogenesis.181 A nontumorigenic hamster cell line became tumorigenic in association with the loss of a suppressor gene and concomitant with the onset of angiogenic activity. The nontumorigenic line secreted high levels of an angiogenesis inhibitor, a truncated form of TSP-1, that decreased by about 96% in the tumorigenic cells.182 TSP-1 was shown to be regulated by the wild-type tumor suppressor p53 in fibroblasts and in mammary epithelial cells.183, 184 Loss of p53 function in the transformed derivatives of these cells dramatically decreased the level of the angiogenesis inhibitor. Restoration of p53 upregulated TSP-1 and raised the antiangiogenic activity of the tumor cells. Deletion of TSP-1 led to accelerated growth of breast cancers that arise spontaneously in neu-transgenic mice.185 The demonstration that the switch to an angiogenic phenotype involved a negative regulator of angiogenesis generated by the tumor per se suggested to Folkman a unifying angiogenic mechanism to explain a well-recognized but previously unsolved clinical and experimental phenomenon: the inhibition of tumor growth by the tumor mass. In this phenomenon, “the removal of certain tumors, for example, breast carcinomas, colon carcinomas, and osteogenic sarcomas can be followed by rapid growth of distant metastases”.186, 187 Postoperative chemotherapy was introduced mainly to prevent or delay the growth of secondary metastases. Several studies in terminally ill patients demonstrated the suppression of a secondary tumor by a primary tumor.188 In melanoma, partial spontaneous regression of the primary tumor may be followed by rapid growth of metastases, and when ionizing radiation is employed to regress a small cell lung cancer, distant metastases may undergo rapid growth.173, 189 Once it was demonstrated that a tumor could generate a negative regulator of angiogenesis, then it became clear that a primary tumor, while stimulating angiogenesis in its own vascular bed, could possibly inhibit angiogenesis in the vascular bed of a distant metastasis.182 However, at least two conditions would be necessary: first, the primary tumor (i.e., the first tumor to grow) would need to generate an angiogenic promoter in excess of an inhibitor in its own vascular bed, and, second, the putative inhibitor would need to have a longer half-life in the circulation than the angiogenic promoter. After arriving in the Folkman laboratory in 1991, O’Reilly and his colleagues validated this hypothesis by discovering angiostatin, endostatin, and antiangiogenic antithrombin over the next 8 years.171–173, 190, 191
Angiostatin
Angiostatin is a 38 kDa internal fragment of plasminogen that was purified from the serum and urine of mice bearing a subcutaneous Lewis lung carcinoma that suppressed growth of its lung metastases by inhibiting their angiogenesis (Figure 5).171 Angiostatin is not secreted by tumor cells but is generated through proteolytic cleavage of circulating plasminogen by a series of enzymes released from the tumor cells. At least one of these tumor-derived enzymes, urokinase plasminogen activator (uPA), converts plasminogen to plasmin, whereas a phosphoglycerate kinase from hypoxic tumor cells then reduces the plasmin so that it can be converted to angiostatin by one of several different metalloproteinases.192 Several potential mechanisms of its antiangiogenic activity have been reported. These include induction of endothelial apoptosis193–195; suppression of endothelial cell migration induced by plasmin binding to αvβ3-integrin196, 197; inhibition of HGF-induced signaling via c-MET, Akt, and ERK-1/2198; and downregulation of VEGF expression in tumor cells.199 This implies that angiostatin may act as a direct and an indirect inhibitor of angiogenesis. In support of this, when angiostatin was delivered to mice bearing a variety of tumors, it showed significant efficacy either alone or in combination with endostatin, as a fusion protein of angiostatin and endostatin, or in combination with interleukin-12.199–202
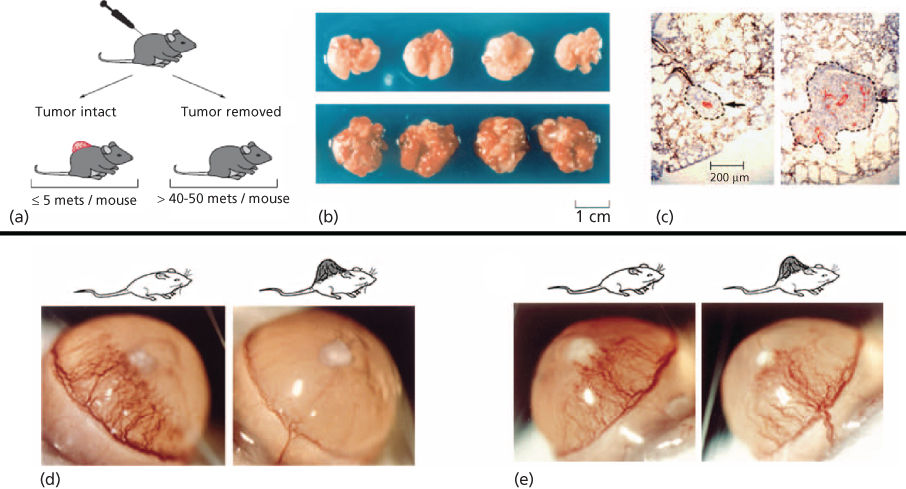
Figure 5 (a) Mice bearing Lewis Lung carcinoma.499 Tumors were resected when tumor size reached 1.5–2 cm2 and the animals were killed after 5 or 15 days. (b) Upper panel: lungs from animals still bearing the primary tumor. Lower panel: lungs removed at the same time from animals in which the tumor had been resected and the animals killed 15 days later. (c) Left panel: microscopic pulmonary metastasis in an animal in which a primary tumor is in place at the same time as the right panel. There is no evidence of angiogenesis as only a single central microvessel stained with antibody to von Willebrand factor. This dormant metastasis is 200 µm in its longest diameter. Right panel: lung metastasis from an animal euthanized 5 days after the primary tumor was removed, showing eight or nine new vessels in an enlarging metastasis. (d) A human prostate carcinoma (LNCaP) growing on the dorsum of an SCID mouse inhibits cornea neovascularization induced by an implanted sustained release pellet of bFGF (80 ng) (right panel). Left panel depicts bFGF-induced corneal neovascularization at 5 days in the absence of a primary tumor. LNCaP prostate cancer generates angiostatin. (e) A human colon cancer that does not produce an angiogenesis inhibitor, growing in an SCID mouse as a control for (d).
Endostatin
A strategy similar to the one that uncovered angiostatin (e.g., suppression of tumor growth by tumor mass) was employed with murine hemangioendothelioma and human small cell lung cancer to discover endostatin and the antiangiogenic conformation of antithrombin-IIIAT.172, 173, 191 Both endostatin and aaAT are generated from larger parent proteins by enzymes released by tumor cells. Endostatin is a 20–22 kDa internal fragment of collagen XVIII.172, 191, 203, 204 It is the first of a group of endogenous angiogenesis inhibitors that are predominantly extracellular proteins, which generally require proteolytic processing to become active.3, 205 Like angiostatin, endostatin appears to have both direct and indirect effects on tumor endothelium, and a number of different mechanisms have been proposed. It is a specific inhibitor of endothelial cell proliferation and migration,206, 207 and blocks the binding of VEGF121 and VEGF165 to the KDR/ Flk-1 receptor and subsequent downstream signaling events.208 Although endostatin does not bind to VEGF,208 it does downregulate VEGF expression in tumor cells.199 In bFGF-treated endothelial cells, endostatin induces endothelial cell apoptosis, in part by activating tyrosine kinase signaling of the Shb adaptor protein.209 Endostatin decreases formation of VEGF-induced microvessels sprouting from aortic rings in vitro210 and inhibits the catalytic activity of MMP-2.211, 212 As a result, endothelial cell invasion is inhibited and tumor cell invasion may also be decreased. Endostatin also inhibits integrin-dependent endothelial cell migration because it binds to α5– and αv-integrins on the endothelial cell surface, in particular α5β1.213 It has been proposed that α5β1-integrin may be a functional receptor for endostatin.34
A wide variety of tumors in many different laboratories have been inhibited by endostatin in mice and rats.214 These include lung adenocarcinoma, thyroid carcinoma, colon carcinoma, leukemia, human non-small cell lung cancer (NSCLC), human pancreatic cancer, human neuroblastoma, breast cancer, and spontaneous pancreatic islet carcinomas.215–222
Tumstatin
Tumstatin (28 kDa) is the NC1 domain fragment of the α3 collagen molecule and exhibits antiangiogenic activity in both in vitro and in vivo assays.223–227 Tumstatin (α3(IV)NC1) binds to endothelial cells via αvβ3-integrin and α6β1-integrin223, 224, 226, 227 and induces apoptosis of proliferating endothelial cells.225 Cell biologic experiments demonstrated that the antiangiogenic activity of tumstatin is dependent on αvβ3-integrin binding to proliferating endothelial cells.223, 227 These experiments support the notion that through the action of endogenous inhibitors such as tumstatin, αvβ3-integrin could also function as a negative regulator of angiogenesis.223, 228–230
Antiangiogenic conformation of antithrombin III
A human small cell lung carcinoma suppressed angiogenesis and tumor growth at remote sites in immunodeficient mice. These cells generated an enzyme in vitro that converted the 58 kDa conformation of circulating antithrombin III to a 53 kDa form of the protein in which the externally configured stressed loop of antithrombin was retracted into the body of the molecule. The 53 kDa “cleaved” form is a specific endothelial inhibitor and a potent angiogenesis inhibitor and has no thrombin-binding activity. Antithrombin III has no antiendothelial or antiangiogenic activity. The enzymes that induce this conformational change have not yet been elucidated. Human pancreatic cancer also generates the 53 kDa cleaved antiangiogenic antithrombin.215
Other pathways regulating angiogenesis
Hypoxia-inducible factor-1
Expression of angiogenic factors including VEGF is positively regulated by hypoxia through the stabilization of the transcription factor hypoxia-inducible factor 1 (HIF-1).231, 232 HIF-1 is a transcription factor comprised of two subunits, HIF-1α and HIF-1β. While HIF-1β is expressed constitutively, expression of HIF-1α is tightly regulated. The stability of HIF-1α is primarily controlled by hypoxia. When oxygen is abundantly present, prolyl hydroxylases modify proline residues 402 and 564 on HIF-1α allowing it to bind the VHL tumor suppressor gene which targets it for degradation.232 Following binding of the HIF-1α and -β subunits, HIF-1 transverses to the nucleus and modulates the expression of numerous genes involved in angiogenesis, cell survival, invasion, and glucose metabolism.232 Indeed, HIF-1α is thought to be the key regulator of potent proangiogenic factors including VEGF.
Although initially thought to be primarily regulated by hypoxia, recent studies have revealed a number of nonhypoxic regulators including receptor tyrosine kinases such as EGFR,233 the PI3K/AKT/mTOR pathway, and metabolic pathways including the tricarboxylic acid (Krebs) cycle (reviewed in Ref. ). Alterations in these pathways have been shown to contribute to inherited cancer syndromes, highlighting their role in carcinogenesis. For example, germline mutations in the VHL gene underlying von Hippel-Lindau disease lead to a markedly elevated risk of developing renal cell carcinoma (RCC), hemangioblastomas of the CNS, and other tumor types. The VHL protein encoded by this gene is part of a protein complex that targets HIFs for degradation.234 Sporadic mutations in the VHL gene also occur in sporadic clear-cell RCC. A second syndrome, hereditary leiomyomatosis and RCC, results from mutations in the fumarate hydratase (FH) gene.236 FH is a mitochondrial protein involved in the TCA cycle. Although the mechanism(s) by which FH mutations promote tumorigenesis is still under investigation, it appears that loss of FH function results in an increase in HIFs by causing a buildup of intracellular fumarate, which inhibits the enzymes (HIF hydroxylases, also known as EGLNs) that hydroxylate HIFs and target them for VHL-mediated degradation. A third hereditary syndrome, tuberous sclerosis complex, is caused by mutations in the tuberous sclerosis complex, resulting in elevated HIFs via the mTOR pathway.237, 238 Several other syndromes have also been identified that resulted in elevated levels of HIFs and downstream HIF-regulated gene products, causing a “pseudohypoxic” state.235, 237 The observations that multiple inherited cancer syndromes involve pathways that converge on HIFs support the hypothesis that dysregulation of the pathways regulating angiogenesis is likely to play a role in driving early tumorigenesis. Several drugs targeting HIFs are currently in clinical development.
Oncogenes as regulators of angiogenesis
It is widely established that activation of proto-oncogenes can induce tumorigenesis. In tissue culture, expression of activated oncogenes increases cell proliferation and decreases apoptosis.239 While these changes contribute to tumorigenesis by altering the equilibrium between cell proliferation and apoptosis, there is considerable evidence that this alone is not sufficient to produce expansive tumor growth.240, 241 Rather, tumors must also acquire an adequate vascular supply to grow beyond 1–2 mm in diameter. In support of this concept, published reports have demonstrated that transfection of tumor cells with oncogenes results in enhanced production of proangiogenic molecules,242 and the in vivo growth of oncogene-driven tumors can be restricted with angiogenesis inhibitors.243 In lung cancer patients, for example, mutations in K-Ras, p53, and EGFR are among the oncogenes that have been linked to angiogenesis. Oncogenes shown to play a role in regulating angiogenic factors include:
Ras
Ras is one of the most commonly activated oncogenes, occurring in 17–25% of all human tumors.244 In tissue culture studies, transfection of transformed murine endothelial cells with the Ras oncogene results in elevated production of VEGF, and treatment of these cells with the PI3K inhibitor, wortmannin, abrogates VEGF expression, indicating that mutated Ras regulates VEGF expression in a PI3K-dependent manner.245 K-Ras gene mutations were positively associated with high VEGF expression in human NSCLC specimens246 and other disease types. In a transgenic model of melanoma, downregulation of the Ras oncogene in a melanoma driven by doxycycline-inducible Ras led to massive apoptosis of microvascular endothelium in the tumor bed starting within 6 h. Tumor cells began to die days later, and large tumors had completely disappeared by 12 days.247
p53
In addition to its role in the regulation of cell cycle and apoptosis, emerging data indicates that p53 indirectly promotes tumor vascularization by altering the expression of proangiogenic and antiangiogenic molecules. In tissue culture studies, fibroblasts expressing wild-type p53 secrete high levels of the antiangiogenic glycoprotein, TSP-1. However, loss of wild-type p53 and expression of the mutant form result in diminished TSP-1 mRNA and protein. Immunohistochemical evaluation of 73 NSCLC clinical specimens revealed a strong statistical association between p53 nuclear localization and microvessel count.248 Additionally, in an analysis of 107 NSCLC patients, p53 was determined to be significantly associated with VEGF expression and microvessel count.249 It is likely that loss of wild-type p53 enhances tumor cell expression of additional proangiogenic factors in NSCLC. Wild-type p53 has been demonstrated to promote Mdm2-mediated ubiquitination and degradation of HIF-1α.250 The loss of wild-type p53 is associated with elevated levels of HIF-1α in tissue culture and augments hypoxia-induced VEGF expression.250
Myc
Myc is a pleiotropic transcription factor overexpressed in many cancer types, which plays a role in regulating angiogenesis, inflammation, and many other processes. Myc activity has been shown to be regulated by RAS pathway activation,242 which in turn regulates angiogenesis at least in part through effects on TSP-1. Myc has also been shown to interact with the HIF-1α pathway to induce angiogenesis by an hypoxia-independent mechanism251 and regulate the recruitment of mast cells required for myc-driven tumorigenesis in a transgenic mouse model.252
EGFR family
EGFR is a member of the erbB family of receptor tyrosine kinases, which also includes HER2/Neu, HER3 (ErbB3), and HER4 (ErbB4). An expanding body of evidence indicates that activation of EGFR leads to enhanced production of proangiogenic molecules. Initial experiments using prostate cancer cell lines demonstrated that stimulation of tumor cells with EGF elevated HIF-1α expression.253 EGF has been shown to increase VEGF production in some tumor cell lines254, 255 and, conversely, treatment of tumor cells with EGFR inhibitors can decrease VEGF expression in various tumor types.233, 255–257 In NSCLC cell lines, EGF activates HIF-1α and induces expression of the chemokine receptor CXCR4 in tissue culture.258 Moreover, in an immunohistochemical study of 172 NSCLC patients, expression of EGFR was associated with HIF-1α positivity,259 and EGFR mutations causing constitutive receptor activation led to upregulated HIF-1α protein and increased VEGF levels.260
Like EGFR, HER2/Neu has also been shown to play a role in regulating angiogenesis. Blockade of HER2 using the monoclonal antibody trastuzumab (Herceptin) has been shown to block the production of multiple angiogenic factors and induce vessel normalization and regression in a murine model of human breast cancer, and enhance the effects of VEGF pathway blockade.261, 262
Therapeutic approaches to targeting tumor vasculature
Angiogenesis inhibitors versus vascular targeting agents
Angiogenesis is the formation of new vessels from preexisting vasculature. Angiogenesis inhibitors are therefore typically targeted at the early stages in this process, including endothelial sprouting and survival mechanisms, which are often VEGF dependent. Vascular targeting agents (VTAs), also known as vascular disrupting agents (VDAs), differ from angiogenesis inhibitors in that they target established abnormal tumor vasculature.263 VTAs can induce rapid collapse of tumor vasculature, and their effects on normal vasculature can cause a host of side effects including acute coronary syndromes, thrombophlebitis, and tumor pain. None of these agents are currently in routine clinical use for cancer but several have undergone clinical testing. Vadimezan (ASA404) initially demonstrated positive results in combination with chemotherapy as treatment for patients with lung cancer in a phase II trial.264 Unfortunately, these findings were not validated in a subsequent phase III study evaluating vadimezan in combination with carboplatin/paclitaxel as first-line treatment in NSCLC, as no significant differences were demonstrated in overall survival (OS) between vadimezan and placebo arms [median OS 13.4 vs 12.7 months, respectively (hazard ratio; HR = 1.01; p = 0.535)]. Given the lack of clinical benefit, the development of vadimezan in lung cancer has been discontinued by Novartis.265 ABT-751 is another VDA with preclinical anticancer activity, which has failed to demonstrate an improvement in progression-free survival (PFS) in a phase I/II study in combination with docetaxel as second-line treatment for NSCLC.266
Antiangiogenic effects of chemotherapy and other therapeutic agents
Multiple preclinical studies have suggested that several “classical” chemotherapeutic agents may also have potent antiangiogenic or vascular-targeting effects, which may be enhanced by low dose, frequent dosing schedules (metronomic dosing).267–269 Several of these regimens are undergoing clinical evaluation. It also appears that certain chemotherapy agents, particularly taxanes and vinca alkaloids, may have relatively more potent antiangiogenic effects than other drugs,269 which may help explain why there are differences in the degree of enhancement observed when antiangiogenic agents such as bevacizumab are added to chemotherapy. This prompted further examination of a wide variety of drugs which were initially thought to target primarily tumor cells. Many of these were subsequently found to also have antiangiogenic effects, prompting the term “accidental antiangiogenics”.270
Targeting VEGF pathway
VEGF is the prototypic member of a family of structurally related, homodimeric growth factors that includes PlGF, VEGF-B, VEGF-C, VEGF-D, and VEGF-E. As described earlier, VEGF family members bind to a family of transmembrane receptor tyrosine kinases that include VEGFR-1 (Flt-1), VEGFR-2 (KDR, Flk-1), and VEGFR-3 (Flt-4) (Figure 3). The effects of VEGF or VEGFR on vascular permeability and endothelial proliferation, migration, and survival are thought to be primarily mediated by VEGFR-2, while VEGFR-3 is primarily expressed on lymphatic endothelium (reviewed in Ref. ). Agents targeting the VEGF pathway include monoclonal antibodies that bind the ligand (i.e., bevacizumab) or block the receptor (ramucirumab). In addition, many small molecule receptor tyrosine kinase inhibitors (RTKIs) have been developed to target critical signaling pathways involved in angiogenesis. These RTKs, which include vatalanib (PTK787), vandetanib (ZD6474), sunitinib (SU11248), axitinib (AG-013736), and nintedanib (BIBF 1120), have been the focus of many preclinical and clinical studies. Because of the structural similarity of the different receptor tyrosine kinase domains, RTKIs typically inhibit multiple receptors such as PDGFR, c-KIT, FGFR, and Axl in addition to VEGFR. These multitargeting agents have demonstrated higher anticancer activity compared to agents with single targets. Furthermore, they are orally available, and thus more convenient for patients. Conversely, the off-target effects may have additive toxicities. These profiles of receptor specificity for each inhibitor, as well as their pharmacokinetics and potency for receptor inhibition, are likely to be the key determinants of their clinical activity. Representative agents targeting the VEGF pathway that are currently FDA approved are listed in Table 2.
Table 2 Approved VEGF pathway inhibitors
| Type | Agent | Target | Approval |
| Monoclonal antibody | Bevacizumab (Avastin) | VEGF-A | FDA approved for CRC, breast cancer, NSCLC, platinum-resistant ovarian carcinoma, and late-stage cervical cancer |
| IMC-1121B Ramucirumab | VEGFR-2 extracellular domain | FDA approved for advanced gastric or gastro-esophageal junction adenocarcinoma; metastatic NSCLC | |
| Soluble decoy receptor | VEGF Trap (Aflibercept) | VEGF-A, VEGF-B, and PIGF | FDA approved for metastatic colorectal cancer |
| RTKIs | Vandetanib | VEGFR-2, EGFR, and RET | FDA approved for progressive medullary thyroid carcinoma |
| Sorafenib | VEGFR-2 and 3, PDGFR-β, Flt-3, c-Kit, and B-Raf | FDA approved for renal cell, hepatocellular cancers, and metastatic differentiated thyroid carcinoma | |
| Sunitinib | VEGFR-1,2, PDGFR, c-Kit, RET, and Flt-3 | FDA approved for renal cell carcinoma, gastrointestinal stromal tumors, and neuroendocrine tumors of the pancreas | |
| AG-013736 (axitinib) | VEGFR-1,2,3 and PDGFR | FDA approved for RCC | |
| Lenvatinib | VEGFR-2,3 | FDA approved for thyroid cancer | |
| BIBF 1120 (Nintedanib) | VEGFR-1,2,3, PDGFR, and FGFR-1/3 | Approved by FDA and EU for idiopathic pulmonary fibrosis, Approved by EU for NSCLC | |
| GW786034 (pazopanib) | VEGFR2 | FDA approved for RCC, and soft tissue sarcoma |
AML, acute myelogenous leukemia; CRC, colorectal cancer; EGFR, epidermal growth factor receptor; FDA, Food and Drug Administration; EU, European Union; GBM, glioblastoma multiforme; NSCLC, non-small cell lung cancer; PlGF, placental growth factor; PDGFR, platelet-derived growth factor receptor; RCC, renal cell carcinoma; RTKI, small molecule receptor tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Combinations of antiangiogenics with chemotherapy: mechanisms for enhanced antitumor activity
Preclinical and clinical studies have demonstrated that antiangiogenic therapy improves the outcome of cytotoxic therapies.271, 272 This finding is paradoxical. It was initially expected that targeting the tumor vasculature would drastically diminish the delivery of oxygen and therapeutics to the solid tumor, producing hypoxia that would cause many chemotherapeutics, as well as radiation, to be less effective.271, 272 Tumor vasculature is known to be structurally and functionally abnormal, with tortuous, highly permeable vessels. Blood flow within these intratumoral vessels is nonuniform. Proliferating tumor cells compress blood and lymphatic vessels resulting in a microenvironment typified by interstitial hypertension (elevated hydrostatic pressure outside the blood vessels), acidosis, and hypoxia.273, 274 This deficient vascular network and interstitial hypertension impair drug delivery to tumor cells. Moreover, hypoxia renders tumor cells resistant to radiation and several cytotoxic agents, and increases genetic instability selecting for tumor cells that have a greater metastatic potential. In addition to the direct effects on tumor cells, hypoxia leads to vascular abnormalization by signaling via PHD2 in tumor endothelial cells275 and, along with the low pH within the tumor microenvironment, weakens the cytotoxic functions of tumor infiltrating immune cells. Collectively, the abnormal vasculature within solid tumors creates a significant barrier to delivery and efficacy of cancer therapeutics.
One proposed mechanism to explain the enhancement in efficacy of chemotherapy by antiangiogenic therapy is that these agents have the potential to “normalize” tumor vessels. In animal models of cancer, inhibition of VEGF signaling results in a vasculature network that more closely resembles vessels within normal tissue. This “normalized” vasculature is less leaky, is less dilated, and has less tortuous vessels with a more normal basement membrane and increased pericyte coverage. Concurrent with these changes in vascular morphology, within the tumor there is a decrease in IFP, increased oxygenation, and improved delivery of concurrently administered chemotherapeutics (Figure 6).276–284 Evidence from a Phase I/II clinical trial in locally advanced rectal carcinoma patients receiving bevacizumab and chemotherapy (with radiation) corroborates preclinical findings. Bevacizumab treatment was associated with a decrease in tumor IFP and an increase in mature, pericyte-covered vessels.285, 286
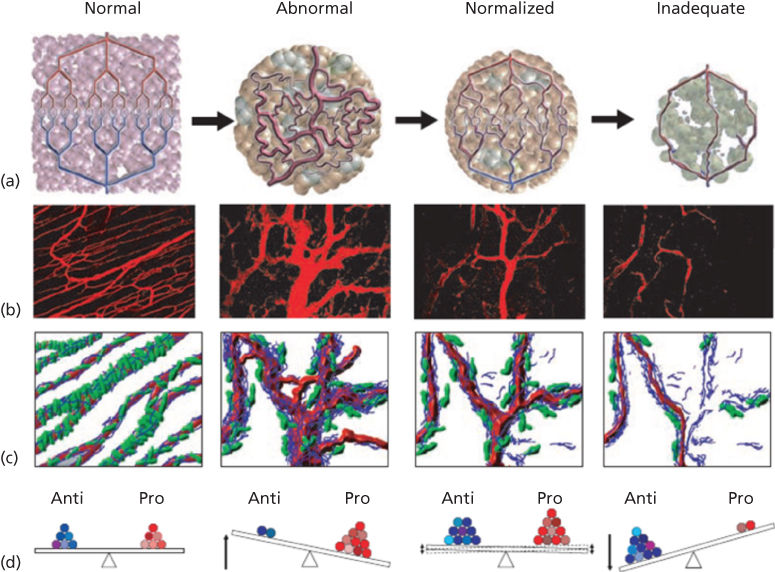
Figure 6 Changes in tumor vasculature during treatment with antiangiogenic agents.271 (a) The tumor vascular network is structurally and functionally abnormal. Antiangiogenic therapies might initially improve both the structure and the function of tumor vessels. Continued or aggressive antiangiogenic regimens may eventually result in a vascular supply that is inadequate to support tumor growth. (b) Vascular normalization due to inhibition of VEGFR2. On the left is a two-photon image depicting normal blood vessels in skeletal muscle; subsequent representative images show human colon carcinoma vasculature in mice at day 0, day 3, and day 5 after treatment with a VEGR2-specific antibody. (c) Diagram illustrating the concomitant changes in basement membrane (blue) and pericyte (red) coverage during vessel normalization. (d) These changes in the vasculature may reflect changes in the balance of pro- and antiangiogenic factors in the microenvironment.
Antiangiogenic agents in combination with radiotherapy
A growing body of evidence suggests that antiangiogenic therapy may enhance the efficacy of radiotherapy for solid tumors. There are several proposed mechanisms through which this enhancement may occur. First, radiotherapy may act by “normalizing” the disorganized and hyperpermeable vasculature in tumors.271, 287 Vessel normalization would permit a more effective delivery of oxygen to tumor tissue, resulting in a reduction in tumor hypoxia and augmenting radiation-induced cytotoxicity in part by increasing the formation of oxygen-free radicals. This reduction in hypoxia may be transient, however, as prolonged use of antiangiogenic agents may eventually also reduce the “normalized” vessels within tumors resulting in an inadequate intratumoral vascular supply, such that the tumor would again be hypoxic with reduced radiosensitivity. This concept is supported by mouse xenografts showing the existence of a period of time (“normalization window”) during which radiation therapy used in conjunction with an antiangiogenic agent is most effective.283, 288
Antiangiogenic therapy may act to enhance the antiendothelial effects of radiotherapy. While it was initially assumed that the antitumor effect of radiotherapy was due to direct action on tumor cells, more recent evidence has demonstrated that radiotherapy also induces endothelial cell apoptosis.288–290 Other studies have confirmed these results.291 Thus, the exact mechanism(s) of interaction between antiangiogenic agents and radiation remains unclear.
Nevertheless, several preclinical studies demonstrate that antiangiogenic agents can synergize with or potentiate the effects of radiotherapy.292–296 Blockade of VEGF signaling on endothelial cells may render the tumor-associated vasculature more sensitive to radiotherapy. In addition, radiation induces VEGF expression, which contributes to radioresistance by blocking radiation-induced endothelial cell apoptosis.297, 298
One possible cause for concern in testing combinations of antiangiogenic therapy and radiotherapy is the observation from preclinical studies that at least some of the toxicities of radiotherapy such as intestinal radiation damage may also be due to endothelial apoptosis.299 In lung cancer patients treated with the combination of bevacizumab, chemotherapy, and radiotherapy, tracheoesophageal fistulas have been observed.300 Clearly additional studies will be needed to assess the feasibility and efficacy of these combinations.
Clinical advances in the use of antiangiogenic therapy
The hypothesis that tumor angiogenesis could serve as a target for cancer therapy is now strongly supported by results of a number of randomized phase III clinical trials across multiple different tumor types. Bevacizumab, a monoclonal antibody targeting VEGF, is now a standard therapy for metastatic colorectal, non-small cell lung, and other tumor types301, 302; VEGFR TKIs such as sunitinib, pazopanib, axitinib, and sorafenib are now approved for RCC and other diseases.303, 304 These advances, coupled with our increased understanding of the biological pathways underlying tumor angiogenesis and the development of improved agents for targeting these pathways, have led to a dramatic increase in the number of clinical trials employing antiangiogenic therapy either alone or in combination with other therapeutic modalities. Currently, the majority of these agents target the VEGF pathway because of its role as a key regulator of tumor angiogenesis.
VEGF pathway inhibitors as anticancer therapy: clinical experience. Clinical trials of one of the earliest VEGF pathway inhibitors, bevacizumab, began in 1997. When used as monotherapy for the treatment of advanced solid tumors, the clinical activity of these agents, as judged by objective tumor responses, has generally been low with the exception of RCC. For example, no partial or complete remissions were observed in 25 patients treated in a phase I trial of bevacizumab.305 Furthermore, in 243 previously treated patients with colorectal cancer, objective response rates (ORR) were 3% for bevacizumab monotherapy, 9.2% for FOLFOX4 chemotherapy (fluorouracil, oxaliplatin, leucovorin), and 21.8% for the combination. Low response rates have also been reported for VEGFR TKIs when used as single agents as discussed below. For this reason, VEGF pathway inhibitors have often been developed as part of combination regimen with chemotherapy or other targeted therapeutics. Major findings in the clinical development of VEGF pathway inhibitors for several common tumor types are reviewed below.
Renal cell cancer
One tumor type for which VEGF pathway inhibitors are particularly useful, even when used as monotherapy, is metastatic RCC. These tumors are often marked by inactivation of the von Hippel-Lindau gene leading to overexpression of VEGF and other angiogenesis mediators (reviewed in Ref. 306). In randomized studies, bevacizumab, sorafenib, and pazopanib have been shown to significantly prolong time to progression compared to placebo controls.307–309 Sunitinib and axitinib also demonstrated substantial antitumor activity in metastatic RCC, with 25–45% ORR in phase III testing.304, 310, 311
Cytokine therapies (interferon-α- and interleukin-2-based therapies) had long been the mainstay of therapy for RCC, so it was rational to evaluate anti-VEGF therapy in the context of such agents. Phase III trials examining four small molecule RTKIs of VEGF receptors (sorafenib, sunitinib, pazopanib, and axitinib) and monoclonal anti-VEGF therapy (bevacizumab) have been reported, and consistently favor the use of anti-VEGF therapy compared to interferon.
Trials of anti-VEGF therapy versus cytokine therapy
Motzer and colleagues compared the use of sunitinib (at a dose of 50 mg orally daily for 4 weeks, followed by 2 weeks without treatment) or interferon α (9 MU subcutaneously three times weekly) in patients with previously untreated, metastatic RCC.304 The primary endpoint of PFS was significantly improved in patients receiving sunitinib (11 vs 5 months, p < 0.001). Patients receiving sunitinib also experienced higher response rates, longer OS, and improved quality of life.310
Trials of cytokine therapy with or without anti-VEGF therapy
Escudier et al.312 and the Cancer and Leukemia Group B performed placebo-controlled Phase III trials comparing the use of interferon α-2a to interferon-α-2a plus bevacizumab in patients with previously untreated metastatic RCC.313 PFS was significantly longer in patients receiving the combined biologic approach. There was a trend to improved OS favoring the bevacizumab-containing treatments.314, 315 More than 50% of the patients received second-line therapy including VEGF-targeted agents, potentially affecting the survival difference.315, 316
Trials of anti-VEGF therapy following cytokine therapy
Escudier et al.303 performed a Phase III trial of sorafenib (400 mg twice daily) versus placebo in patients who had progressed following first-line cytokine therapy for advanced RCC. Median PFS was 5.5 months in the sorafenib group and 2.8 months in the placebo group (p < 0.01). Analysis of OS showed that sorafenib reduced the risk of death (HR, 0.88; 95% CI, 0.74–1.04; p = 0.146).317 Although this benefit was not statistically significant, censoring of placebo-assigned patients who crossed over to sorafenib uncovered a significant result (17.8 vs 14.3 months, respectively; HR = 0.78; 95% CI, 0.62–0.97; p = 0.0287).
Therapy following initial anti-VEGF treatment
The demonstration of a beneficial effect for anti-VEGF therapy in RCC has fundamentally altered the treatment landscape for this disease. Nevertheless, anti-VEGF therapy is not curative and only modestly prolongs survival, leaving room for substantial therapeutic improvement. Patients progressing on one VEGF-targeting agent may still respond to another.318–320 Whether this cross-sensitivity is a function of the promiscuity of RTKIs, of differential pharmacokinetics, or of varying affinities for VEGF receptors is unknown.
mTOR inhibition in RCC
Two trials321, 322 examined the role of mTOR inhibition in patients with advanced RCC. While mTOR has several biologic roles, one is as a downstream effector of VEGF signaling. In a trial comparing the mTOR inhibitor temsirolimus versus interferon α versus the combination of temsirolimus plus interferon α as front-line therapy, Hudes et al.321 demonstrated the superiority of temsirolimus to interferon α, with a significant improvement in OS (10.9 months vs 7.3 months, p = 0.008); the combination arm was not superior to interferon monotherapy. In addition, the temsirolimus group had fewer serious adverse events than the interferon α group. In a placebo-controlled randomized Phase III trial in the post-VEGF TKI setting, the mTOR inhibitor everolimus significantly prolonged PFS when compared to placebo (4 vs 1.9 months, p < 0.001).322 OS was not significantly different, perhaps due to the fact that crossover was allowed for patients progressing on placebo. Though toxicities were generally mild, significant increases in severe (grade 3 or 4) toxicities were seen for stomatitis, p = 0.03; infections, p = 0.03; hypercholesterolemia, p = 0.03; hyperglycemia, p < 00001; lymphopenia, p = 0.002; and hypophosphatemia, p = 0.01. No prospective data is available that directly compares everolimus with a VEGF RTKI in patients whose disease has progressed on prior treatment with a VEGF inhibitor, but, OS was shown inferior for temsirolimus as compared with sorafenib (12.3 vs 16.6 months, respectively; stratified HR, 1.31; 95% CI, 1.05–1.63; two-sided p = 0.01) after disease progression on sunitinib.323 Moreover, one randomized phase II clinical trial suggested that everolimus is inferior to sunitinib regarding PFS, particularly when used in first line.324 There is no evidence to support the use of combinations of agents with different mechanisms of action as compared to sequential use, and toxicity may be increased, as shown for the combination of bevacizumab with temsirolimus (compared to bevacizumab and interferon).325
Antiangiogenic therapy as adjuvant therapy in RCC
The advent of multiple active antiangiogenic agents led to the development of several adjuvant trials in RCC. Early results of one, E2805, in which patients were randomly assigned to 1 year of treatment with sorafenib (400 mg twice daily), sunitinib (50 mg/day for 4 of every 6 weeks), or placebo, indicated that neither sorafenib nor sunitinib prolonged time to disease recurrence after surgery (5.6 years with either sorafenib or sunitinib vs 5.7 years with placebo).326
Prediction of therapeutic benefit in RCC
It has been established that relatively high VEGF levels are associated with worse tumor stage and grade, performance status, and overall prognosis.317, 327, 328 Moreover, in a phase III trial of sorafenib versus placebo, patients with VEGF in the highest concentration quartile obtained greater relative benefit from sorafenib than those with lower concentrations.327 However, studies addressing whether VEGF is a predictive marker for identifying RCC patients who are likely to benefit from VEGF-targeted therapies have yielded inconsistent results.329 A recent study in samples from two subsequent phase II and III studies identified interleukin (IL)-6 as predictive of PFS benefit from pazopanib versus placebo.330 Unfortunately, no biomarkers that are predictive of differential benefit between available and active drugs in RCC have yet been validated.
Future directions in RCC
While FGF and VEGF receptor inhibition with dovitinib failed to improve PFS relative to sorafenib in third line after progression to VEGF and mTOR inhibition,331 coinhibition of other pathways of potential escape to VEGF signaling blockade such as cMET remains promising. Given very promising results of novel immunotherapeutic agents such as checkpoint inhibitors in RCC, multiple clinical trials are now evaluating their combination with angiogenesis inhibitors.
Colorectal cancer
Advanced colorectal cancer represented the first human cancer in which a Phase III trial demonstrated clinical benefit, and to this day represents among the best studied of human solid tumors with regard to antiangiogenic therapy. In 2004, results of a phase III, randomized, placebo-controlled study were reported comparing standard IFL chemotherapy (irinotecan, fluorouracil, and leucovorin) with or without bevacizumab in patients with previously untreated metastatic colorectal cancer.301 Patients treated with IFL plus bevacizumab had a significantly longer OS and PFS as well as a higher ORR. This trial provided definitive evidence that the addition of an angiogenesis inhibitor to chemotherapy could prolong survival and, based on the results of this trial, bevacizumab received approval from the US Food and Drug Administration (FDA) for use in combination with fluorouracil-containing chemotherapy as first-line treatment for metastatic colorectal cancer.
Subsequent studies have built upon this original finding. In a trial of previously treated patients with colorectal cancer, patients treated with bevacizumab combined with FOLFOX4 chemotherapy had a prolonged survival compared to those treated with FOLFOX4 alone,332 although the magnitude of this benefit (2.1 months) appeared to be smaller than that observed in the first-line trial (4.7 months), presumably related to the more advanced nature of the disease.
First-line studies of bevacizumab or placebo in combination with either FOLFOX or XELOX failed to demonstrate an improvement in OS (21.3 vs 19.9 months) despite improvements in PFS (9.4 vs 8.0 months).333 The mechanism for the discrepancy between this study and the pivotal IFL study has been attributed to the different cytotoxic backbone or practice patterns of treatment until progression of disease versus regimen interruptions for cumulative oxaliplatin toxicities. Several studies have evaluated the role of bevacizumab after an initial course of oxaliplatin- and fluoropyrimidine-based first-line chemotherapy. These “maintenance” regimens have demonstrated the benefit of bevacizumab when combined with fluoropyrimidines. The CAIRO-3 randomized study demonstrated that continued bevacizumab either alone or in combination with capecitabine provided a survival benefit compared to observation alone.
Cohort studies had suggested a benefit from continued VEGF inhibition after progression on first-line therapy with bevacizumab. The TML study evaluated second-line therapy with FOLFIRI or FOLFOX (as determined by the treating physician) with or without bevacizumab for patients who had previously progressed on first-line bevacizumab-containing regimen. The study demonstrated a modest but statistically significant survival advantage of 1.4 months, resulting in the addition of bevacizumab continuation to treatment guidelines.
A similar study was conducted with ziv-aflibercept, a fusion protein utilizing the extracellular domains of VEGFR1 and VEGFR2, thereby providing inhibition of both VEGF-A and PlGF. Patients who had been previously treated with an oxaliplatin regimen (which may have included bevacizumab) were randomized to FOLFIRI with bevacizumab or placebo.334 The primary endpoint of OS was met with a 1.4 month improvement (13.5 months vs 12.1 months), with an improvement in PFS and response rate. Grade 3 and 4 diarrhea and stomatitis were substantially higher in the ziv-aflibercept arm, consistent with the established role for PlGF in mucosal lining repair.335 The result of these two studies is the addition of new treatment options for second-line antiangiogenic therapy in combination with chemotherapy for patients who have previously progressed on bevacizumab-based first-line regimens.
Biomarker analyses of colorectal cancer trials have been largely unavailing. Expression levels of VEGF-A, thrombospondin, and microvessel density performed in a subset analysis of the original Hurwitz trial were not associated with clinical outcome.336
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


