Chemical carcinogenesis
Lorne J. Hofseth, PhD  Ainsley Weston, PhD
Ainsley Weston, PhD  Curtis C. Harris, MD
Curtis C. Harris, MD
Overview
Human exposure to chemical carcinogens can result in cancer. What dictates this outcome is relatively predictable but highly variable. Factors governing the outcome include type of exposure, amount of exposure, time of exposure, and genetic makeup of the host (i.e., humans). The latter is comprised of variations in single nucleotides within genes (e.g., single nucleotide polymorphisms in deoxyribonucleic acid–repair genes), as well as the metabolomic, proteomic, microbiomic, transcriptomic, and epigenomic background of the individual. It is becoming increasingly clear that these endpoints can be modified by chemical carcinogens and that inflammatory load influences outcome. To this end, the past decade has seen an explosion of extremely sensitive and highly accurate technology. Linking this technology to the rapid development of bioinformatics has enabled us to begin merging the totality of lifetime exposure (exposome) with the totality of metabolomic, proteomic, microbiomic, transcriptomic, epigenomic, and other “omic” profiles. We feel optimistic that the next decade will bring the development of tools to identify an individual’s weighted risk signature as a biomarker for cancer risk and develop a personalized and precise approach to cancer chemoprevention and treatment.
Human chemical carcinogenesis is a multistage process that results from carcinogen exposure; usually in the form of complex chemical mixtures, and often encountered in the environment or through our lifestyle and diet (Tables 1 and 2). A prominent example is tobacco smoke, which can cause cancers at multiple sites with the highest risk being that of lung cancer. Although most chemical carcinogens do not react directly with intracellular components, they are activated to carcinogenic and mutagenic electrophiles by metabolic processes evolutionarily designed to clear the body of toxins and to modify endogenous compounds. Electrophilic chemical species are naturally attracted to nucleophiles like deoxyribonucleic acid (DNA) and protein; and genetic damage results through covalent bonding to DNA. Once internalized, carcinogens are subject to competing processes of metabolic activation and detoxification, although some chemical species can act directly. There is considerable variation among the human population in these competing metabolic processes, as well as the capacity for repair of DNA damage and cellular growth control. This is the basis for inter-individual variation in cancer risk and is a reflection of gene–environment interactions, which embodies the concept that heritable traits modify the effects of chemical carcinogen exposure.55, 56 Such variations in constitutive metabolism, DNA repair, and cellular growth control contribute to the relative susceptibility of individual members of the population to chemical exposures. For example, only 10% of tobacco smokers develop lung cancer, albeit that tobacco use accounts for other fatal conditions, including cardiovascular disease, emphysema, and chronic obstructive pulmonary disorder (COPD). Within the conceptual framework of multistage carcinogenesis, the primary genetic change that results from a chemical–DNA interaction is termed tumor initiation.13, 14, 57 Thus, initiated cells are irreversibly altered and are at a greater risk of malignant conversion than are normal cells. The epigenetic effects of tumor promoters facilitate the clonal expansion of the initiated cells.58, 59 Selective, clonal growth advantage causes a focus of pre-neoplastic cells to form. These cells are more vulnerable to tumorigenesis because they now present a larger, more rapidly proliferating, target population for the further action of chemical carcinogens, oncogenic viruses, and other cofactors. Additional genetic and epigenetic changes continue to accumulate.58, 59 The activation of oncogenes, and the inactivation of tumor suppressor and DNA-repair genes, leads to genomic instability or the mutator phenotype and an acceleration in the genetic changes taking place.29, 60, 61 This scenario is followed by malignant conversion, tumor progression, and metastasis. The underlying molecular mechanisms that govern chemical carcinogenesis are becoming increasingly understood, and the insights generated are assisting in the development of better methods to investigate human cancer risk and susceptibility. The results of such studies are intended to mold strategies for prevention and intervention. Moreover, insights into the normal operations of so-called gatekeeper genes,62 like the tumor suppressor TP53, have provided an opportunity to develop new, targeted, therapeutic approaches.
Table 1 Selected examples of human chemical carcinogenesis
| Organ system (specific pathology) | Chemical carcinogen | Co-carcinogen |
| Lung (small and nonsmall cell) | Tobacco smoke | Asbestos |
| Metals: As, Be, Cd, Cr, Ni | — | |
| BCME | — | |
| Diesel exhaust | — | |
| Pleural mesothelium | Asbestos | Tobacco smoke |
| Oral cavity | Smokeless tobacco | — |
| Betel quid | Slaked lime [Ca(OH)2] | |
| Esophagus | Tobacco smoke | Alcohol |
| Nasal sinuses | Snuff | Powdered glass |
| Isopropyl alcohol | — | |
| Skin (scrotum) | Cutting oil | — |
| Coal soota | — | |
| Liver (angiosarcoma) | Aflatoxin B1 | HBV, HCB |
| Vinyl chloride | Alcohol | |
| Bladder | Aromatic amines (e.g., 4-ABP and benzidine) | — |
| Aromatic amines from tobacco smokeb | — | |
| ALL | Benzene | — |
| Lymphatic and hematopoietic malignancies | Ethylene oxide | — |
4-ABP, 4-aminobiphenyl; ALL, acute lymphoblastic leukemia; BCME, bischloromethyl ether; HBV, hepatitis B virus; HCV, hepatitis C virus.
a Early report of occupational chemical carcinogenesis from 225 years ago.
b Strong circumstantial evidence.1
A comprehensive treatise on the evaluation of the carcinogenic risk of chemicals to humans can be found in the ongoing International Agency for Research on Cancer monograph program initiated in 1971.
Table 2 Some landmark discoveries in chemical carcinogenesis
| Year | Event/discovery/finding | References |
| 3000 BC | First written description of cancer (breast) in the Edwin Smith Papyrus | 2 |
| 1500 BC | Egyptians treat tumors with chemicals (arsenic) | 2 |
| 1620 | Thomas Venner warns about immoderate use of tobacco | 3, 4 |
| 1742 | Hermann Boerhaave and Jean Astruc link inflammation to cancer | 3, 5, 6 |
| 1775 | Percival Pott describes association between soot exposure and scrotal cancer in chimney sweeps | 7 |
| 1863 | Rudolf Virchow: cancers tend to occur at sites of chronic inflammation | 8 |
| 1879 | Harting and Hesse: lung cancer is an occupational disease of miners | 9 |
| 1909 | First chemotherapy drug (an arsenobenzene analog, compound 606, or salvarsan) treats syphilis | 10 |
| 1910 | Viruses found to cause cancer | 11 |
| 1932 | Female hormones (estrogen) cause breast cancer in mice | 12 |
| 1941 | Two-stage initiation and promotion of cancers by chemical carcinogens proposed | 13, 14 |
| 1950 | Tobacco exposure linked to lung cancer | 15, 16 |
| 1956 | Nitrosamines cause cancer | 17 |
| 1956 | Evidence that enzymes can be activated by chemical carcinogens | 18 |
| 1962, 1963 | Evidence that chemical carcinogens can methylate DNA and proteins | 19, 20 |
| 1964–1968 | Aflatoxin B1, a fungal toxin, is carcinogenic in rats, binds to DNA and is carcinogenic in humans | 21–23 |
| 1968 | DNA-repair defects linked to cancer | 24 |
| 1971 | “Two-hit” theory of cancer causation in the Rb gene, and tumor suppressor gene discovery | 25 |
| 1971 | Epigenetic mechanisms for carcinogenesis proposed | 26 |
| 1973 | Ames assay identified to test mutagenicity of chemical carcinogens | 27 |
| 1974 | Evidence that chemical carcinogens are activated to form DNA adducts in human tissues | 28 |
| 1974 | Mutator phenotype concept identified | 29 |
| 1976 | Inter-individual variation in the binding of chemical carcinogens to DNA and gene–environment interactions | 30 |
| 1973 | Oncogene discovery | 31, 32 |
| 1977 | Cancer stem cells identified | 33 |
| 1979 | p53, the most frequently altered gene in human cancer, discovered | 34, 35 |
| 1981 | First quantitative estimation of the contribution of the environment and genetics to carcinogenesis | 36 |
| 1982 | Formal structure/model of molecular epidemiology introduced | 37 |
| 1983 | Single nucleotide polymorphisms can guide carcinogenesis | 38 |
| 1988 | Chemical carcinogens cause site-specific mutagenesis | 39 |
| 1990 | Sequential mutations in colon cancer introduced | 40 |
| 1991 | p53 identified as a tumor suppressor gene | 41 |
| 1991 | Identification of selective mutations in p53 (mutational hotspots) associated with specific environmental and chemical carcinogens | 42, 43 |
| 2001 | Draft sequence of the human genome | 44, 45 |
| 2002 | MicroRNA alterations in cancer | 46 |
| 2002 | Gene expression signatures predict prognosis | 47 |
| 2005 | Concept of the exposome (the entirety of exposure to which an individual is subjected, from conception to death) is introduced | 48 |
| 2006 | Genetic landscape of two human cancer types defined (colon and breast) | 49 |
| 2006 | MicroRNA signatures predict prognosis and survival | 50 |
| 2006 | FDA approves HPV vaccine [human papillomavirus quadrivalent (types 6, 11, 16, 18) vaccine, recombinant] | 51 |
| 2011 | National Research Council (United States) committee on A Framework for Developing a New Taxonomy of Disease introduces precision medicine | 52 |
| 2012 | Victor Velculescu: detection of chromosomal alterations in the circulation of cancer patients | 53 |
| 2012 | Use of integrative personal omics profile to distinguish between healthy and diseased states at an individual level | 54 |
Multistage carcinogenesis
Carcinogenesis can be divided conceptually into four steps: tumor initiation, tumor promotion, malignant conversion, and tumor progression (Figure 1, Box 1). The distinction between initiation and promotion was recognized through studies involving both viruses and chemical carcinogens.13, 14, 57 This distinction was formally defined in a murine skin carcinogenesis model in which mice were treated topically with a single dose of a polycyclic aromatic hydrocarbon (PAH) (i.e., initiator), followed by repeated topical doses of croton oil (i.e., promoter),13 and this model has been expanded to a range of other rodent tissues, including bladder, colon, esophagus, liver, lung, mammary gland, stomach, and trachea.59 During the last 65 years, the sequence of events comprising chemical carcinogenesis has been systematically dissected and the paradigm increasingly refined, and both similarities and differences between rodent and human carcinogenesis have been identified.63 Carcinogenesis requires the malignant conversion of benign hyperplastic cells to a malignant state, and invasion and metastasis are manifestations of further genetic and epigenetic changes.64 The study of this process in humans is necessarily indirect and uses information from lifestyle or occupational exposures to chemical carcinogens. Measures of age-dependent cancer incidence have shown, however, that the rate of tumor development is proportional to the sixth power of time, suggesting that at least four to six independent steps are necessary.65 Partial scheduling of specific genetic events in this process has been possible for some cancers. Examples of sequential genetic and epigenetic changes that occur with the highest probability are those found in the development of lung cancer66 and colon cancer.67 Recent advances in sequencing technology have allowed us to identify the genomic landscape of many tumors. In common solid tumors such as those derived from the colon, breast, brain, or pancreas, an average of 33–66 genes display subtle somatic mutations that can alter their protein products.68 Certain tumor types display many more or many fewer mutations than average. Melanomas and lung tumors, for example, contain approximately 200 nonsynonymous mutations per tumor; a reflection of the involvement of potent mutagens [ultraviolet (UV) light and cigarette smoke, respectively] in the pathogenesis of these tumor types.68
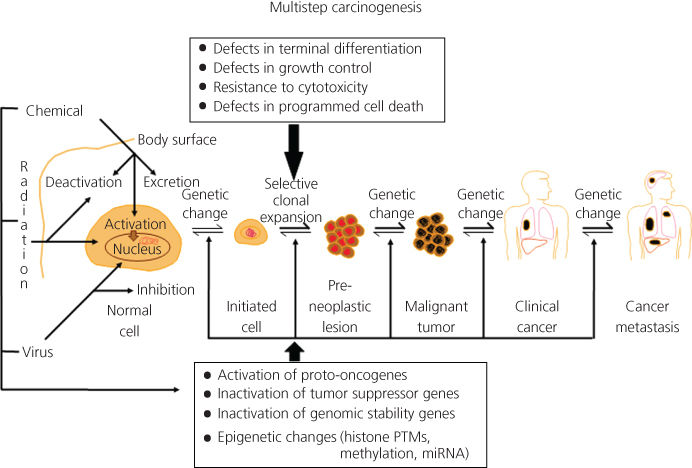
Figure 1 Multistep chemical carcinogenesis can be conceptually divided into four stages: tumor initiation, tumor promotion, malignant conversion, and tumor progression. The activation of proto-oncogenes and the inactivation of tumor suppressor genes are mutational events that result from covalent damage to DNA caused by chemical exposures. The accumulation of mutations, and not necessarily the order in which they occur, constitutes multistage carcinogenesis. During these stages, progressive epigenetic changes are also occurring due to chemical exposure.
Tumor initiation
Earlier concepts of tumor initiation indicated that the initial changes in chemical carcinogenesis are irreversible genetic damage. However, recent data from molecular studies of pre-neoplastic human lung and colon tissues implicate epigenetic changes as an early event in carcinogenesis. DNA methylation of promoter regions of genes can transcriptionally silence tumor suppressor genes.69 In a broad sense, then, chemical carcinogens can be divided into genotoxic [e.g., benzo(a)pyrene or B(a)P; generally considered to act at the initiation stage] and nongenotoxic agents [e.g., 3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) or 12-O-tetradecanoylphorbol-13-acetate (TPA); generally considered to act in the promotion stages]. These nongenotoxic or epigenetic agents (Figure 1) neither do not induce mutations nor do they induce direct DNA damage in the target organ. They modulate cell growth and death and exhibit dose-response relationships between exposure and tumor formation. Although the exact mechanism(s) of action of these agents on neoplastic cell formation are yet to be determined, changes in gene expression and cell growth parameters appear to be critical, and nongenotoxic compounds exhibit temporal and threshold characteristics frequently requiring chronic treatment for carcinogenicity.
Overall, most human cancers are caused by two to eight sequential alterations that develop over the course of 20–30 years.68 For mutations to accumulate, they must arise in cells that proliferate and survive the lifetime of the organism. A chemical carcinogen causes a genetic error by modifying the molecular structure of DNA that can lead to a mutation during DNA synthesis. Most often, this is brought about by forming an adduct between the chemical carcinogen or one of its functional groups and a nucleotide in DNA (the process by which this occurs for the major classes of chemical carcinogens is discussed in detail in “Carcinogen Metabolism”). In general, a positive correlation is found between the amount of carcinogen–DNA adducts that can be detected in animal models and the number of tumors that develop.70, 71 Thus, tumors rarely develop in tissues that do not form carcinogen–DNA adducts. Carcinogen–DNA adduct formation is central to theories of chemical carcinogenesis, and it may be a necessary, but not a sufficient, prerequisite for tumor initiation (the concept of so-called nongenotoxic carcinogens is also explored in “Carcinogen Metabolism”). DNA adduct formation that causes either the activation of a proto-oncogene or the inactivation of a tumor suppressor gene can be categorized as a tumor-initiating event (see Sections titled “Tumor progression” and “Oncogenes and tumor suppressor genes”).
Tumor promotion
Tumor promotion comprises the selective clonal expansion of initiated cells. Because the accumulation rate of mutations is proportional to the rate of cell division, or at least the rate at which stem cells are replaced, clonal expansion of initiated cells, produces a larger population of cells that are at risk of further genetic changes and malignant conversion.64, 68 Tumor promoters are generally nongenotoxic, are not carcinogenic alone, and often (but not always) are able to mediate their biologic effects without metabolic activation. These agents are characterized by their ability to reduce the latency period for tumor formation after exposure of a tissue to a tumor initiator, or to increase the number of tumors formed in that tissue. In addition, they induce tumor formation in conjunction with a dose of an initiator that is too low to be carcinogenic alone. Croton oil (isolated from Croton tiglium seeds) is used widely as a tumor promoter in murine skin carcinogenesis, and the mechanism of action for its most potent constituent, TPA, which occurs via protein kinase C activation, is arguably the best understood among tumor promoters.72 Chemicals or agents capable of both tumor initiation and promotion are known as complete carcinogens [e.g., B(a)P and 4-aminobiphenyl]. Identification of new tumor promoters in animal models has accelerated with the sophisticated development of model systems designed to assay for tumor promotion. Furthermore, ligand-binding properties can be determined in recombinant protein kinase C isozymes that are expressed in cell culture.73 Chemicals, complex mixtures of chemicals, or other agents that have been shown to have tumor-promoting properties include dioxin, TPA, TCDD, benzoyl peroxide, macrocyclic lactones, bromomethylbenzanthracene, anthralin, phenol, saccharin, tryptophan, dichlorodiphenyltrichloroethane (DDT), phenobarbital, cigarette-smoke condensate, polychlorinated biphenyls (PCBs), teleocidins, cyclamates, estrogens and other hormones, bile acids, UV light, wounding, abrasion, and other chronic irritation (i.e., saline lavage).74
Malignant conversion
Malignant conversion is the transformation of a pre-neoplastic cell into one that expresses the malignant phenotype. This process requires further genetic changes. The total dose of a tumor promoter is less significant than frequently repeated administrations, and if the tumor promoter is discontinued before malignant conversion has occurred, premalignant or benign lesions may regress. Tumor promotion contributes to the process of carcinogenesis by the expansion of a population of initiated cells, with a growth advantage, that will then be at risk for malignant conversion. Conversion of a fraction of these cells to malignancy will be accelerated in proportion to the rate of cell division and the quantity of dividing cells in the benign tumor or pre-neoplastic lesion. In part, these further genetic changes may result from infidelity of DNA synthesis.75 The relatively low probability of malignant conversion can be increased substantially by the exposure of pre-neoplastic cells to DNA damaging agents,76 and this process may be mediated through the activation of proto-oncogenes and inactivation of tumor suppressor genes.
Box 1 Multistage carcinogenesis
Multistage carcinogenesis involves four stages:
- 1. Tumor initiation: The initial changes to normal cells that occur early in chemical carcinogenesis and involve irreversible genetic mutation(s) (genotoxic initiation) or epigenetic changes (nongenotoxic initiation) so that they are able to form tumors.
- 2. Tumor promotion: The selective clonal expansion of a population of initiated cells, causing additional genetic changes with a growth advantage that will then be at risk for malignant conversion. Tumor promoters are generally nongenotoxic, cannot drive tumorigenesis alone, and require repeat exposure over time.
- 3. Malignant conversion: The transformation of a preneoplastic cell into one that expresses a malignant phenotype. The probability of malignant conversion increases through additional genetic changes, continued exposure of preneoplastic cells to DNA damaging agents, and may be mediated through the activation of proto-oncogenes and inactivation of tumor suppressor genes.
- 4. Tumor progression: The expression of the malignant phenotype and the tendency of malignant cells to acquire more aggressive characteristics over time. A prominent characteristic of the malignant phenotype is the propensity for genomic instability and uncontrolled growth. During this process, further genetic and epigenetic changes can occur, again including the activation of proto-oncogenes and the functional loss of tumor suppressor genes.
Tumor progression
Tumor progression comprises the expression of the malignant phenotype and the tendency of malignant cells to acquire more aggressive characteristics over time. Also, metastasis may involve the ability of tumor cells to secrete proteases that allow invasion beyond the immediate primary tumor location. A prominent characteristic of the malignant phenotype is the propensity for genomic instability and uncontrolled growth.77 During this process, further genetic and epigenetic changes can occur, again including the activation of proto-oncogenes and the functional loss of tumor suppressor genes. Frequently, proto-oncogenes are activated by two major mechanisms: in the case of the ras gene family, point mutations are found in highly specific regions of the gene (i.e., the 12th, 13th, 59th, or 61st codons), and members of the myc, raf, HER2, and jun multigene families can be overexpressed, sometimes involving amplification of chromosomal segments containing these genes. Some genes are overexpressed if they are translocated and become juxtaposed to a powerful promoter, for example, the relationship of bcl-2 and immunoglobulin heavy chain gene promoter regions in B-cell malignancies. Loss of function of tumor suppressor genes usually occurs in a bimodal fashion, and most frequently involves point mutations (caused by DNA adducts, errors in DNA replication, and errors in DNA repair) in one allele and loss of the second allele by a deletion (caused by a recombinational event, chromosomal nondisjunction, or hypermethylation). These phenomena confer to the cells a growth advantage as well as the capacity for regional invasion, and ultimately, distant metastatic spread. Despite evidence for an apparent scheduling of certain mutational events, it is the accumulation of these mutations, and not the order or the stage of tumorigenesis in which they occur, that appears to be the determining factor.68 Recent evidence from microarray expression analysis of human cancers supports an alternative, and not mutually exclusive, mode of tumor progression. Gene expression profiles of a primary cancer and its metastases are similar, indicating the molecular progression of a primary cancer is generally retained in its metastases.78
Over the last decade, sequencing efforts have identified genomic landscapes of common forms of human cancer. Vogelstein and colleagues recently defined these landscapes as consisting of a small number of “mountains” (genes altered in a high percentage of tumors) and a much larger number of “hills” (genes altered infrequently).68 Approximately 140 genes that when altered by intragenic mutations can drive tumorigenesis. A typical tumor contains two to eight of these “driver gene” mutations that control cell fate, cell survival, and genomic maintenance. Other “passenger mutations” confer no selective growth advantage. Overall, the identification of specific genes and their function in primary cancers and metastases have clinical implications in molecular diagnosis of primary cancers and targeted therapeutic strategies for personalized medicine.
Epigenetics and chemical carcinogenesis
Epigenetics describes a change in gene activity without a change in the DNA sequence.79 Well-described mechanisms involved in epigenetics include DNA methylation and histone modification; each of which alters how genes are expressed without altering the underlying DNA sequence. Another mechanism (described in detail later) also falling under the definition of “epigenetic” is the effects of microRNA (miRNA; Figure 2, Box 2) on carcinogenesis. Both nongenotoxic and genotoxic chemical carcinogens impact these epigenetic processes. At the cellular level, exposure to environmental factors may leave an epigenomic signature that can be exploited in discovering new biomarkers for risk assessment and cancer prevention.80, 81
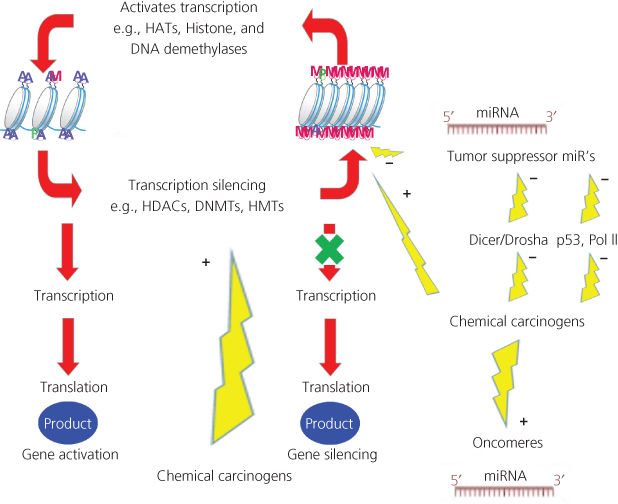
Figure 2 Epigenetic impact of chemical carcinogens. Chemical carcinogens can affect the activity of the different enzymes involved in epigenomics. Ac: acetyl; Me: methyl; DNMT: DNA methyltransferase; HAT: histone acetyltransferase; HDAC: histone deacetylase; HMT: histone methyltransferase.
Box 2 Epigenetics
Epigenetics describes a change in gene activity without a change in the DNA sequence. It involves changes in DNA methylation [hypomethylation/hypermethylation caused by carcinogens modifying methylation enzymes (e.g., DNMTs)], histone tail modification [acetylation, methylation, citrullination, ubiquitination, sumoylation, and phosphorylation caused by carcinogens modifying enzymes involved in such changes (e.g., HDACs)], and small noncoding RNA’s (e.g., miRNAs caused by genetic abnormalities, modulation of biogenesis machinery, and/or epigenetic mechanisms).
DNA methylation is catalyzed by a family of DNA methyltransferases (DNMTs). In somatic mammalian cells, DNA methylation occurs at cytosine residues of CpG dinucleotides; in embryonic stem cells, DNA methylation occurs at both CpG and non-CpG sequences. DNMT expression, and therefore indirect control of DNA methylation, is controlled by DNMT3L, lymphoid-specific helicase, miRNAs, and piwi-interacting RNAs.82 It is becoming increasingly clear that many chemical carcinogens are involved in DNA methylation. For example, the tobacco-specific carcinogen, 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone (NNK), induces DNMT accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients.83 As well, B(a)P and many others not only genotoxic, but also nongenotoxic chemical carcinogens have been shown to induce aberrant methylation patterns.82, 84 Several mechanisms may explain aberrant methylation patterns due to chemical carcinogens. As an example, cigarette smoke may alter DNA methylation by (1) inducing DNA damage, stimulating recruitment of DNMT1; (2) the ability of nicotine to down-regulate DNMT1 mRNA and protein expression; (3) modulating expression and activity of DNA-binding factors, such as Sp1; and (4) inducing hypoxia, which increases HIF1α, leading to the up-regulation of methionine adenosyltransferase 2A85 (Figure 2).
Chromatin is the complex of DNA wrapped around histone octamers, consisting of four different histones, H2A, H2B, H3, and H4. Gene expression changes with histone post-transcriptional modification (e.g., acetylation, phosphorylation, and methylation) in the N-terminal tail region. Enzymes that regulate histone modification include histone deacetylases (HDACs), histone acetyltransferases (HATs), and histone methyltransferases (HMTs). Chemical carcinogens can alter the activity of such enzymes. For example, challenging cells with B(a)P induces early enrichment of the transcriptionally active chromatin markers histone H3 trimethylated at lysine 4 (H3K4Me3) and histone H3 acetylated at lysine 9 (H3K9Ac), and reduces association of DNMT1 with the long interspersed nuclear element-1 (L1) promoter. These changes are followed by proteasome-dependent decreases in cellular DNMT1 expression and sustained reduction of cytosine methylation within the L1 promoter CpG island.86 Similarly, long-term exposure of immortalized bronchial epithelial cells (HBEC-3KT) to low doses of tobacco-related carcinogens leads to oncogenic transformation, increased HDAC expression, cell-cycle independent increased DNMT1 stability, and DNA hypermethylation.87
miRNA’s in chemical carcinogenesis
miRNA expression is regulated by a growing list of factors, including inflammatory cytokines, free radicals, and chemical carcinogens.88–91 The specific effects of chemical carcinogens on each miRNA in vitro and in vivo have been described in detail elsewhere, so the reader is guided to that source.91 For the purposes of this chapter, we will provide an overview of miRNAs, examples of key miRNAs affected by chemical carcinogens, and the mechanisms by which this occurs.
miRNAs are nonprotein coding small RNAs of about 22 base pairs and regulate mRNA stability and translation into proteins.92 miRNA genes are regulated by transcription factors including by the p53 tumor suppressor protein93 and are involved in carcinogenesis including tumor invasion and metastasis.94 miRNAs are produced by processing of a hairpin precursor by an enzyme called dicer, and the mature miRNA incorporates into a protein complex (the RNA-induced silencing complex—RISC), where it acts as a guide to find complementary target RNAs. Binding of RISC to the target RNA causes degradation of the target or blocks translation—in either case, preventing production of the encoded protein. The alteration of miRNA expression by chemical exposure or during inflammation potentially has enormous consequences, because of the large number of genes each miRNA regulates. Each organism encodes hundreds of miRNAs, and these affect many key biological processes, including cell proliferation, differentiation, survival, and metabolism. In the 15 or so years since miRNAs were discovered,95–97 it has become clear that post-transcriptional control of gene expression by miRNAs provides an important and widely used level of gene regulation. In humans, it is estimated that about 60% of all protein-coding genes are regulated by a miRNA.98, 99 Because miRNAs are highly specific, potent suppressors of gene expression; and aberrant regulation of miRNA expression is associated with cancer (and many other diseases), the potential value of these small RNAs as therapeutic agents is now widely recognized.100–104 Not surprisingly, miRNAs are also clinical biomarkers associated with diagnosis, prognosis, and therapeutic outcome of cancer.105, 106 Because miRNA levels change upon exposure to environmental and endogenous chemicals, it is possible that miRNAs can be used for biomonitoring purposes. This is supported by the understanding that miRNAs are released from the target tissues into the blood stream; and therefore, their analysis is feasible via noninvasive sampling (e.g., urine and feces).107–109
Oncogenic miRNAs (OncomiRs)
In 2002, the Croce group reported the first link between miRNA and the pathogenesis of cancer; specifically, miR-15a and miR-16-1 were observed to be downregulated in B-cell chronic lymphocytic leukemia (CLL) patients.46 Since this seminal observation, many of the known miRNA genes were shown to reside in genomic regions that are commonly associated with cancer.110 To this end, miRNAs themselves are now known to act as tumor suppressors and oncogenes (oncomiRs); and environmental carcinogens alter miRNA expression by down-regulating suppressor miRNA and up-regulating oncomiRs.
OncomiRs are miRNA that have a causal role in driving cancer phenotypes. As discussed throughout this chapter, cancer represents the summation of extensive genetic and epigenetic abnormalities found in genes involved in diverse biological pathways. Even with this complexity, certain cancers can be dependent on a single, powerful oncogene, and abrogation of this oncogene can reverse the malignant phenotype. Targeting the Philadelphia chromosome with inhibitors of the BCR-ABL kinase domain in chronic myelogenous leukemia is a classic, successful example of this approach.111, 112 Herceptin and its success in treating Her2+ breast cancer is another example.113, 114 It is also known that single oncogenes can affect miRNA expression, exemplified by Myc, which can induce the oncomiR miRNA-17/92 cluster, and negatively regulate tumor suppressor miRNA’s, such as let-7 and miRNA-29.115, 116 Thus, the potential to target oncomiRs is appealing due to their capacity to control many cancer pathways. One way is to identify the exogenous and/or endogenous (e.g., free radicals) chemicals leading to an increase in oncomiR expression or imbalance of oncomiRs. miR-21 is one key oncomiR, up-regulated in a number of human cancers.90, 117 It has been shown to be up-regulated by exposure to cancer-causing agents such as arsenite,118 reactive oxygen species (ROS),118 hydrogen peroxide,119 UV irradiation,120 TPA,121 and nitric oxide from NOS2.122 In addition, absence of miRNA-21 results in suppressed skin tumorigenesis induced by 7,12-dimethylbenz[a]anthracene (DMBA)/TPA. miR-21 targets include multiple tumor suppressor genes, such as phosphatase and tensin homolog (PTEN) and tropomyosin α-1 (TPM1) as well as DNA mismatch repair genes, mutS homolog 2 (MSH2) and mutS homolog 6 (MSH6).117 Other oncomiRs, including miRNA’s-17/92, -20, -106, -107, -141, -146, -155, -181, -200, -221/222, and -373, have all been shown to be a target of chemical carcinogens (e.g., BPDE, DMBA),92, 123 in that their expression is increased by these agents. This gives us initial insight into chemicals to target, as well as molecular oncomiR targets/signatures for protection against cancers associated with these chemicals.
Suppressor miRNAs
Many forms of cancer are associated with misregulation of miRNA expression and, indeed, miRNA expression profiles are now used as one way to classify tumors.105, 124–126 Often, tumor cells show a decrease in the level of certain miRNAs that function as tumor suppressors [for reviews, see refs 100, 127–130]. Each tumor suppressor miRNA is thought to regulate the expression of a broad spectrum of genes that work in different cancer pathways. Although only a subset of the direct targets of each tumor suppressor miRNA is currently known, it is clear that the overall impact of losing a tumor suppressor miRNA is to promote one or more aspects of carcinogenesis. In several cases, experiments using animal models have demonstrated that loss of a particular tumor suppressor miRNA can drive tumorigenesis. In these cases, tumorigenesis is suppressed when the missing miRNAs are added back.129, 131–133 miRNA-34a, which is a component of the p53 transcriptional network134 and regulates cancer stem cell survival,135, 136 is a good example of this, and so far, arguably, appears to be the furthest in development for use in clinical trials.127, 136 miRNA-34a is down regulated in cancer and has been demonstrated to block the initiation and/or inhibit the growth of tumors in animal models of different kinds of cancer when levels are restored.137–142 Thus, miRNA replacement therapy appears quite promising for treating cancer. However, as is the case for other therapies based on small RNAs, delivery of the RNA to target sites is a critical barrier to implementation.130, 133, 143, 144 A variety of systems have been developed to overcome these challenges. Many methods make use of commercially synthesized molecules produced with novel chemical modifications designed to improve stability.144–146 In addition, synthetic RNAs are generally introduced as a duplex rather than as the mature single-stranded miRNA, a feature that not only promotes stability, but also triggers mammalian defense pathways that recognize double-stranded RNA. In many cases, the chemically modified RNA is conjugated to another molecule (e.g., cholesterol) or encapsulated into nanoparticles to overcome barriers to cellular uptake of the RNA.123, 141 Downsides here include the following: (1) the specificity and functionality of the modified molecule may be altered from that of the natural miRNA, resulting in decreased therapeutic potential; (2) adverse side effects can be induced, including stimulation of immune response, toxicity due to packaging components, and activation of the complement pathway; and (3) increased expense. Furthermore, delivery strategies involving expression of small RNA precursors via adenoviral- or lentiviral-based vectors are often used in animal models but are not deemed safe enough for therapies in humans. Thus, although researchers have met with some success in systemic delivery of therapeutic small RNAs, many challenges remain, including safety concerns, efficacy, and economic feasibility.
There are multiple examples of changes in (mostly decreases in) suppressor miRNA levels by chemical carcinogens. The full list has been reviewed recently, so the reader is referred to the reference.91 Highlights include suppression of the suppressor miRNA’s let-7 family and miR-34b in lungs of cigarette smoke-exposed mice. Interestingly, the expression of the let-7 family was restored 1 week after smoking cessation. However, the recovery was incomplete for a limited array of miRNAs, including miRs-34b, -345, -421, -450b, -466, and -469. Thus, it appears that miRNAs mainly behave as biomarkers of effect and that exposure to high dose, lasting for an adequate period of time, is needed to trigger the cigarette-smoke-related carcinogenesis process associated with miRNA changes.147
Mechanisms of chemical carcinogen-induced changes in miRNA expression
Changes in miRNA levels by environmental carcinogens appear to be reversible at low doses and short periods of exposure, but irreversible with higher doses and long periods of exposure; and that irreversible miRNA changes are predictive of the future appearance of cancer.91, 147 The mechanisms of how chemical carcinogens lead to an imbalance and changes in miRNA expression appear to involve genetic abnormalities, modulation of the biogenesis machinery, and/or epigenetic mechanisms92, 115, 116 (Box 2). Genetic abnormalities include chromosomal rearrangements, genomic amplifications, deletions, and mutations.92 This was demonstrated in the first studies identifying miRNA’s in tumorigenesis. The miRNA-15/16-1 cluster was shown to be deleted in CLL, with subsequent reduced accumulation.46 Germline mutation was also associated with reduced accumulation of this cluster.148 The miRNA biogenesis machinery can be altered through multiple mechanism, each resulting in altered miRNA levels. For example, mutations can occur in key biogenesis enzymes (Dicer and Drosha) and/or their complexes,149 resulting in aberrant miRNA production. Indeed, it has been shown that a series of chemical carcinogens (e.g., PAHs, heterocyclic compounds, nitrosamines, morpholine, ethylnitrosourea, benzene derivatives, hydroxyl amines, and alkenes) interfere with miRNA maturation by binding Dicer.91, 149 Although the effects of environmental chemical carcinogens on the miRNA machinery have been described in detail recently,91 they can be summed up by three mechanisms. First, in response to DNA damage, the p53/miRNA interconnection can modify the expression of miRNA genes in the nucleus. Second, electrophilic metabolites of environmental carcinogens can bind to nucleophilic sites of miRNA precursors thus forming miRNA adducts that cannot access the catalytic pockets of Dicer in the cytoplasm. Third, metabolites of environmental carcinogens can bind to Dicer in the proximity of miRNA catalytic sites thus blocking maturation of miRNA precursors91 (Figure 2). It remains, however, unclear to why oncomiR’s are upregulated, which will be an important mechanism to delineate for a better understanding of chemical carcinogenesis and chemoprevention. To this end, there are ongoing studies identifying specific small molecule inhibitors of oncomiR biogenesis.150
Because miRNA expression is also altered by nongenotoxic carcinogens,91 there are additional mechanisms modulating expression outside of the DNA damage response pathway. Epigenetic mechanisms (e.g., hypermethylation of tumor suppressor miRNA), as well as direct effects on the transcriptional machinery and post-transcriptional modifiers, play a role. For example, hypermethylation of miRNA-124a in colon cancer results in the up-regulation of the oncogene, Cdk6 kinase and the phosphorylation of the tumor suppressor, pRb.151 CpG island hypermethylation of additional tumor suppressor miRNAs have been shown, and the list is growing.152–155 miRNA’s have also been shown to target and alter the activity of DNMT and other enzymes involved in epigenetic modulation (HDACs and histone acetyl transferases)156–159 (Figure 2). Carcinogens, in turn, can play an integral part in this process. For example, nickel sulfide, which is a weak mutagen but strong carcinogen, can down-regulate miRNA-152 via promoter-DNA hypermethylation.157 Similarly, ROS inhibit miR-199a and miR-125b expression through increasing the promoter methylation of the miR-199a and miR-125b genes by DNMT1.160
As many miRNAs are transcribed by PolII and associated factors,92, 161, 162 it stands to reason that miRNA levels are altered by an impact of environmental carcinogens on PolII and other factors with resulting altered transcriptional regulation. miR-34a and miR-34b/c, for example, are direct, conserved p53 target genes that mediate induction of apoptosis, cell-cycle arrest, and senescence by p53.163 When p53 is mutated (often by environmental carcinogens164), its’ transcriptional activity is compromised.165 In a similar way, environmental carcinogens affect the activity of PolII.166
Overall, it is clear that chemical carcinogens alter miRNA expression. As with many biological entities, these changes occur initially due to exposure as an adaptive response to the chemical insult. Such mechanisms that miRNA work include the activation of p53, cell-cycle arrest, and apoptotic induction (e.g., by the miR-34 suppressor miRNA family). Proto-oncogene mutation (“initiation”; e.g., k-ras) can induce expression of other suppressor miR’s such as the let-7 family.167 With long-term, extensive exposure, miRNA expression changes can become irreversible. This has been shown with the miR-34 and let-7 family members where their inactivation results in the suppression of p53 and activation of k-ras.91, 168 If miRNA replacement therapy becomes mainstream, this will be a powerful tool for changing and perhaps reversing the carcinogenic process induced by chemical carcinogens. Interestingly, recent studies have found that plant miRNAs consumed by mammals are detected in mammals (including humans), have activity in mammals, and have tumor-suppressive properties.169–171
Gene–environment interactions and inter-individual variation
A cornerstone of human chemical carcinogenesis is the concept of gene–environment interactions (Figure 3, Box 3).56, 172 Potential inter-individual susceptibility to chemical carcinogenesis may well be defined by genetic variations in the host elements of this compound system. Functional polymorphisms in human proteins that have, or may have, a role in chemical carcinogenesis include enzymes that metabolize (i.e., activate and detoxify) xenobiotic substances, enzymes that repair DNA damage, cell surface receptors that activate the phosphorylation cascade and cell-cycle control genes (i.e., oncogenes and tumor suppressor genes that are elements of the signal transduction cascade).
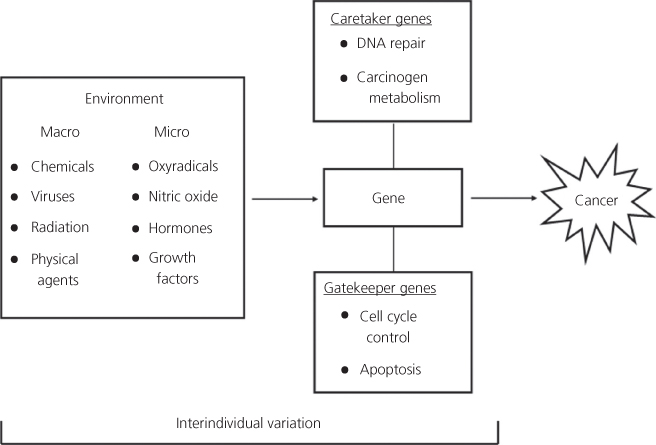
Figure 3 The concept of gene–environment interactions is multifaceted: (1) environmental chemicals are altered by the products of metabolic genes; (2) environmental chemicals disrupt the expression (induce or inhibit) of carcinogen metabolizing genes; and (3) environmental exposures cause changes (mutations) in cancer-related genes. The cancer-related genes have been classified as gatekeeper (e.g., APC) and caretaker (e.g., MSH1 and MLH1) genes. The interaction of these genes with external and internal environmental agents can lead to the derangement of regulatory pathways that maintain genetic stability and cellular proliferation.
Box 3 Gene–environment interactions
Genes identified that modify response to chemical carcinogens include those involved in carcinogen metabolism (e.g., p450’s), DNA repair (e.g., NER), cell signaling, cell cycle, and hormonal regulation.
When chemicals or xenobiotics encounter biologic systems, they become altered by metabolic processes. This is an initial facet of gene–environment interaction. The inter-individual variation in carcinogen metabolism and macromolecular adduct formation arising from such processes was recognized almost 40 years ago.30 The cytochrome P450 (CYP) multigene family is largely responsible for the metabolic activation and detoxification of many different chemical carcinogens in the human environment.173 CYP P450s are phase I enzymes that act by adding an atom of oxygen onto the substrate; they are induced by PAHs and chlorinated hydrocarbons.173 Phase II enzymes act on oxidized substrates and also contribute to xenobiotic metabolism. Some phase II enzymes are methyltransferases, acetyltransferases, glutathione transferases, uridine 5′-diphosphoglucuronosyl transferases, sulfotransferases, nicotinamide adenine dinucleotide (NAD)- and nicotinamide adenine dinucleotide phosphate (NADP)-dependent alcohol dehydrogenases, aldehyde, steroid dehydrogenases, quinone reductases, NADPH diaphorase, azo reductases, aldoketoreductases, transaminases, esterases, and hydrolases. The pathways of activation and detoxification are often competitive, providing yet further potential for individual differences in propensity for carcinogen metabolism to DNA damaging species. This scenario is further complicated by a second facet of gene–environment interaction that leads to enzyme induction or inhibition. In this case, environmental exposures alter gene expression, and genes responsible for carcinogen metabolism can be upregulated or repressed by certain chemical exposures.
A third facet of gene–environment interaction occurs when the chemical alters gene structure (environmentally predisposed abnormal DNA). Once a procarcinogen is metabolically activated to an ultimate carcinogenic form, it can bind covalently to cellular macromolecules, including DNA. This DNA damage can be repaired by several mechanisms.174 Differences in rates and fidelity of DNA repair potentially influence the extent of carcinogen adduct formation (i.e., biologically effective dose) and, consequently, the total amount of genetic damage. The consequences of polymorphisms in genes controlling the cell cycle (serine/threonine kinases, transcription factors, cyclins, cyclin-dependent kinase inhibitors, and cell surface receptors) are much less clear. However, molecular epidemiologic evidence suggests that certain common variants of these types of genes have a role in susceptibility to chemical carcinogenesis.175 The evaluation of polymorphisms as potential biomarkers of susceptibility in the human population is discussed in Weston and Harris.176
Carcinogen metabolism
Inter-individual variation in cancer susceptibility, and, consequently, meaningful human cancer risk assessment, involve determination of inherited host factors as well as exposure assessment. Metabolic polymorphisms have historically been determined by the use of indicator drugs (e.g., caffeine, debrisoquine, dextromethorphan, dapsone, and isoniazid), however, these assays are being replaced by direct genetic assays. This approach has allowed the investigation of diverse host factors for which indicator drugs were not available, and it has been applied to a wide variety of cancers.177–181 Such studies have not only made a key impact in the area of pharmacogenomics,182, 183 but genetic indicators of propensity for carcinogen activation and detoxification, DNA-repair capacity, apoptosis, and cell-cycle control are all features of molecular epidemiologic studies that are complementary to adduct studies because of the implications for a biologically effective dose after exposure.
CYP P450 polymorphisms, involved in carcinogen activation, and glutathione-S transferases, uridine diphosphate (UDP) glucuronosyltransferases, sulfotransferases, and N-acyltransferases, involved in both carcinogen activation and detoxification, could explain variations in cancer susceptibility among the human population. Evidence that absent protection of a functionally intact GSTM1 gene correlates with an increased risk of tobacco-related lung cancer.184 Similarly, the absence of GSTM1 and GSTT1 genes increases the risk of lung cancer because of radon exposure.185 There is a substantial reduction in risk of lung adenocarcinoma associated with genetic polymorphism in CYP2A13, the most active CYP P450 for the metabolic activation of tobacco-specific carcinogen NNK.186 The finding that XRCC1, GSTM1, and COMT polymorphisms was strongly associated with lung cancer risk in smoking women is supportive of an interaction of repair, tobacco smoke metabolism, and estrogen metabolism in this disease and continues the controversy of an increased risk of lung cancer in women.187 Also, UDP glucuronosyltransferases (e.g., UGT1A1, UGT1A9, and UGT2B7) have been implicated in cancers of the head and neck. Persons inheriting reduced activity variants of NAT1 and NAT2 genes, resulting in the slow acetylator phenotype, are at a greater risk of aromatic amine-induced bladder cancer. This may include persons exposed through tobacco smoke inhalation.188, 189 Over the past 25 years, there have been advances in the understanding of carcinogen metabolism, inter-individual variations, and cancer risk. A good review of the existing literature can be found elsewhere.190
PAHs, for example, B(a)P, were the first carcinogens to be chemically isolated.191 They are composed of variable numbers of fused benzene rings that form from incomplete combustion of fossil fuels and vegetable matter, they are common environmental contaminants. PAHs are chemically inert, and require metabolism to exert their biologic effects.191 This multistep process involves the following: initial epoxidation (CYP P450), hydration of the epoxide (epoxide hydrolase), and subsequent epoxidation across the olefinic bond.173, 192 The result is the ultimate carcinogenic metabolite; in the case of B(a)P it is r7, t8-dihydroxy-c9, 10 epoxy-7,8,9,10-tetrahydroxybenzo[a]pyrene (B[a]P-7,8-diol 9,10-epoxide, BPDE).193 The biology of CYP P450 (e.g., CYP1A1) metabolism has been elucidated providing a molecular basis for inducibility and inter-individual variation, and variations in CYP levels among humans have been documented.194 The arene ring of BPDE opens spontaneously at the tenth position, revealing a carbonium ion that can form a covalent addition product (adduct) with cellular macromolecules, including DNA. Several DNA adducts can be formed, the most abundant being at the exocyclic amino group of deoxyguanosine ([7R]-N 2-[10–7β,8a,9a-trihydroxy-7,8,9,10-tetrahydro-benz[a]pyrene}yl]-deoxyguanosine; BPdG). One electron oxidation has been suggested as an alternative pathway of PAH activation, here a radical cation forms at the meso position (L-region). The resulting DNA adducts at the C8 of guanine (B(a)P-6-C8Gua and BP-6-C8dGua), the N7 of guanine and adenine (B(a)P-6-N7Gua and BP-6-N7Ade) likely undergo spontaneous depurination. Firm evidence for the exfoliation of these adducts in urine has been provided.195
Aromatic amines are found in cigarette smoke, diesel exhaust, industrial environments, and certain cooked foods. The compound, 4-aminobiphenyl, is thought to be responsible for bladder cancer among tobacco smokers and rubber industry workers.196 In addition, nitrated PAHs are environmental contaminants that are related to aromatic amines by nitroreduction. Aromatic amines can be converted to an aromatic amide that is catalyzed by an acetyl coenzyme A-dependent acetylation.197 The acetylation phenotype varies among the population. Persons with the rapid acetylator phenotype are at a higher risk of colon cancer (especially in smokers),198 whereas, those who are slow acetylators are at risk of bladder cancer.198 This latter association may result from the fact that activation of aromatic amines by N-oxidation is a competing pathway for aromatic amine metabolism. The N-hydroxylation products when protonated (e.g., in the urinary bladder) are reactive and can cause DNA damage.
An initial activation step for both aromatic amines and amides is N-oxidation by CYP1A2. The reactions of N-hydroxyarylamines with DNA appear to be acid catalyzed, but they can be further activated by either an acetyl coenzyme A-dependent O-acetylase or a 3′-phosphoadenosine-5′phosphosulfate-dependent O-sulfotransferase. The N-arylhydroxamic acids arise from the acetylation of N-hydroxyarylamines or N-hydroxylation of aromatic amides; they are not electrophilic and require further activation. The predominant pathway here occurs through acetyltransferase-catalyzed rearrangement to a reactive N-acetoxyarylamine. Sulfotransferase catalysis forms N-sulfonyloxy arylamides. This complex pathway results in two major adduct types, amides (acetylated) and amines (nonacetylated).
Heterocyclic amines form in food cooking from pyrolysis (>150 °C) of amino acids, creatinine, and glucose. They have been recognized as food mutagens, shown to form DNA adducts and cause liver tumors in primates.199 These compounds are activated by CYP1A2 and their metabolites form DNA adducts in humans. The N-hydroxy metabolites of heterocyclic amines like 2-amino-3-methyl-imidazo-[4,5-f]quinoline (IQ) can react directly with DNA. Enzymic O-esterification of N-hydroxy metabolites plays a key role in activating food mutagens, and because the N-hydroxy metabolites are good substrates for transacetylases, these chemicals may be implicated in colorectal cancer.
Aflatoxins (B1, B2, G1, and G2), metabolites of Aspergillus flavus, contaminate cereals, grain, and nuts. A positive correlation exists between dietary aflatoxin exposure and the incidence of liver cancer in developing countries, where grain spoilage is high. Aflatoxins B1 and G1 have an olefinic double bond at the 8,9-position that can be oxidized by several CYP P450.173, 192 This implies that the olefinic 8,9-bond is the activation site. Further support for this mechanism comes from studies of DNA adducts and the prevalence of TP53 mutations in liver cancer. In people with liver cancer from parts of China and Africa, where food spoilage caused by molds is high, G : C to T : A transversions in codon 249 are frequent.42, 43, 200 This phenomenon is consistent with metabolic activation of aflatoxin B1 and the formation of depurinating carcinogen–deoxyguanosine adducts.
Carcinogenic N-nitrosamines are ubiquitous environmental contaminants and can be found in food, alcoholic beverages, cosmetics, cutting oils, hydraulic fluid, rubber, and tobacco.201 Tobacco-specific N-nitrosamines (TSNs), for example, NNK, are not formed by pyrolysis, which accounts for the highly carcinogenic nature of snuff and chewing tobacco.202 TSNs are not symmetric so both small alkyl adducts and large bulky adducts can be formed; for example, NNK metabolism gives rise to either a positively charged pyridyl-oxobutyl ion or a positively charged methyl ion, both of which are able to alkylate DNA.70, 203 Endogenous nitrosamines form when an amine reacts with nitrate alone or nitrite in the presence of acid. Thus, nitrite (used to cure meats) and L-cysteine, in the presence of acetaldehyde (from alcohol), form N-nitrosothiazolidine-4-carboxylic acid. N-Nitrosodimethylamine undergoes a-hydroxylation, catalyzed primarily by the alcohol inducible CYP2E1, to form an unstable a-hydroxynitrosamine. The breakdown products are formaldehyde and methyl diazohydroxide. Methyl diazohydroxide and related compounds are powerful alkylating agents that can add a small functional group at multiple sites in DNA.
Nongenotoxic carcinogens may function at the level of the microenvironment by dysregulation of hormones and growth factors, or indirectly inducing DNA damage and mutations through the action of free radicals. These chemicals are none or poorly reactive and are resistant to activation through metabolism. They are also characterized by their persistence in biological systems and consequently tend to accumulate in the food chain. However, they can stimulate oxyradical formation by at least three mechanisms: organochlorine species interact with the Ah receptor, which can lead to CYP P450 induction and associated oxyradical formation; interaction with other receptors, like interferon (IFN)-γ, can stimulate elements of the primary immune response and again generate oxyradicals; and agents like asbestos can promote oxyradical formation through interaction with ferrous metal. The resulting oxyradicals can then damage DNA. Some of the so-called “nongenotoxic” carcinogens might more appropriately be considered to be “oxyradical triggers.” Indeed, chronic inflammatory states, which involve oxyradical formation, can also be cancer risk factors.204–208
DNA damage and repair
Another facet of inter-individual variation in risk for cancer from chemical carcinogens is inter-individual differences in the ability to repair damage from chemical carcinogens. DNA damage initiates a complex network of signaling cascades.209 The chemical structure of DNA can be altered by a carcinogen in several ways: the formation of large bulky aromatic adducts, small alkyl adducts, oxidation, dimerization, and deamination. In addition, double- and single-strand breaks can occur. Chemical carcinogens can cause epigenetic changes, such as altering the DNA methylation status that leads to the silencing of specific gene expression.69 A complex pattern of carcinogen–DNA adducts likely results from a variety of environmental exposures, because of the mixture of different chemical carcinogens present.
Many DNA-repair genes have been described recently, and a growing number of polymorphisms have been identified for which molecular epidemiologic studies have provided evidence that genetic variation in these attributes can be a human cancer risk factor; as well as be used to tailor cancer chemotherapy.210, 211 Typically, these types of molecular epidemiological studies initially focus on high-exposure groups such as workers, patients taking therapeutic drugs, and tobacco smokers. Several polymorphisms in DNA-repair genes have now been implicated in tobacco-related neoplasms.212 An interesting theory developed recently is a so-called, “hide-then-hit” hypothesis. This describes the importance of DNA-repair variants in escaping checkpoint surveillance. Only cells with subtle defects in repair capacity arising from low-penetrance variants of DNA-repair genes would have the opportunity to grow and to accumulate the genetic changes needed for cancer formation, without triggering cell-cycle checkpoint surveillance.213
BPDE reacts with the exocyclic (N2) amino group of deoxyguanosine and resides within the minor groove of the double helix, it is typical of PAHs. This adduct, BPdG, is probably the most common, persistent adduct of B(a)P in mammalian systems, but others are possible. Adducts like BPdG are thought to induce ras gene mutations, which are common in tobacco-related lung cancers.214, 215 Aromatic amine adducts are more complex, because they have both acetylated and nonacetylated metabolic intermediates, and they form covalent bonds at the C8, N2, and sometimes O6 positions of deoxyguanosine as well as deoxyadenosine. The major adducts, however, are C8-deoxyguanosine adducts, which reside predominantly in the major groove of the DNA double helix.216
Aflatoxin B1 and G1 activation through hydroxylation of the olefinic 8,9-position results in adduct formation at the N7 position of deoxyguanosine. These are relatively unstable with a half-life of approximately 50 h at neutral pH; depurination products have been detected in urine.217 The aflatoxin B1-N7-deoxyguanosine adduct also can undergo ring opening to yield two pyrimidine adducts; alternately, aflatoxin B1-8,9-dihydrodiol could result, restoring the DNA molecular structure if hydrolysis of the original adduct occurs.218
DNA alkylation can occur at many sites either following the metabolic activation of certain N-nitrosamines, or directly by the action of the N-alkyl ureas (N-methyl-N-nitrosourea) or the N-nitrosoguanidines. The protonated alkyl-functional groups that become available to form lesions in DNA generally attack the following nucleophilic centers: adenine (N1, N3, and N7), cytosine (N3), guanine (N2, O6, and N7), and thymine (O2, N3, and O4). Some of these lesions are known to be repaired (O6-methyldeoxyguanosine), while others are not (N7-methyldeoxyguanosine), which explains why O6-methyldeoxyguanosine is a promutagenic lesion and N7-methyldeoxyguanosine is not.219, 220
Another potentially mutagenic cause of DNA damage is the deamination of DNA-methylated cytosine residues. 5-Methylcytosine comprises approximately 3% of deoxynucleotides. In this case, deamination at a CpG dinucleotide gives rise to a TpG mismatch. Repair of this lesion most often restores the CpG; however, repair may cause a mutation (TpA).221 Deamination of cytosine also can generate a C to T transition if uracil glycosylation and G-T mismatch repair are inefficient.
Oxyradical damage can form thymine glycol or 8-hydroxydeoxyguanosine adducts. Exposure to organic peroxides (catechol, hydroquinone, and 4-nitroquinoline-N-oxide) leads to oxyradical damage; however, oxyradicals and hydrogen peroxide can be generated in lipid peroxidation and the catalytic cycling of some enzymes, as well as environmental sources (e.g., tobacco smoke).204, 222 Certain drugs and plasticizers can stimulate cells to produce peroxisomes, and oxyradical formation is mediated through protein kinase C when inflammatory cells are exposed to tumor promoters like phorbol esters.223 Oxyradicals can contribute to deamination through induction of NO synthase.224
Maintenance of genome integrity requires mitigation of DNA damage. Thus, diminished DNA repair capacity is associated with carcinogenesis, birth defects, premature aging, and foreshortened life span. DNA-repair enzymes act at DNA damage sites caused by chemical carcinogens, and six major mechanisms are known: direct DNA repair, nucleotide excision repair, base excision (BER) repair, nonhomologous end joining (double-strand break repair), mismatch repair, and homologous recombination (HR) (postreplication repair).225, 226
In the presence of nonlethal DNA damage, cell-cycle progression is postponed for repair mechanisms. This highly coordinated process involves multiple genes. A DNA-damage recognition sensor triggers a signal transduction cascade and downstream factors direct G1 and G2 arrest in concert with the proteins operationally responsible for the repair process. Although there are at least six discrete repair mechanisms, within five of them, there are numerous multiprotein complexes comprising all the machinery necessary to accomplish the step-by-step repair function.
Generically, DNA repair requires damage recognition, damage removal or excision, resynthesis or patch synthesis, and ligation. Recent advances have led to the cloning of more than 130 human genes involved in five of these DNA-repair pathways. A list of these genes and their specific functions was published elsewhere.227 These genes are responsible for the fidelity of DNA repair, and when they are defective, the mutation rate increases. This is the mutator phenotype.29, 60, 61 Mutations in at least 30 DNA-repair associated genes have been linked to increased cancer susceptibility or premature aging (Table 3).227 Moreover, the role of common polymorphisms in some of these genes is associated with increased susceptibility in a gene–environment interaction scenario (this is discussed in Weston and Harris176). Indeed, molecular epidemiologic evidence suggests that tobacco-smoking-related lung cancer is associated with a polymorphism in the nucleotide excision repair gene, XPC (ERCC2).228
Table 3 Examples of disease susceptibility and disease syndromes associated with mutations in DNA-repair genes
| Gene | Function | Pathology or cancer |
| Cancer susceptibility | ||
| MMRa | ||
| MLH1 | Damage recognition | HNPCC2b, glioma |
| MLH2 | DNA binding | HNPCC1, ovarian cancer |
| MSH3 | — | Endometrial cancer |
| MSH6 | Sliding clamp | Endometrial cancer, HNPCC1 |
| PMS1 | Damage recognition | HNPCC3 |
| PMS2 | Repair initiation | HNPCC4, glioblastoma |
| NER | ||
| BRCA-1 | Directs p53 transcription toward DNA-repair pathways | Breast cancer, ovarian cancer |
| RB1 | Cell-cycle restriction | Retinoblastoma, breast cancer, and progression osteosarcoma |
| DSB | ||
| BRCA-2 | Regulation of RAD51 | Breast cancer, pancreatic cancer |
| HR | ||
| RAD54 | Helicase | Colon cancer, breast cancer, NHL |
| Other | ||
| TP53 (DSB, NER, HR) | Cell-cycle control; exonuclease; apoptosis; DNA binding | Colon cancer, common somatic defect in human cancer in general; inherited in Li–Fraumeni syndrome and some breast cancers |
| hOgg1 (Various) | Glycosylase | Cancer susceptibility |
| Xeroderma pigmentosum (XP) | ||
| NER | ||
| XPD | DNA helicase | Skin and neurologic, but later onset than XPA |
| XPB | DNA helicase | Skin lesions |
| XPG | Endonuclease | Acute sun sensitivity, mild symptoms; late skin cancer |
| XPC (and BER) | Exonuclease | Mental retardation; skin sensitivity; microcephaly |
| DDB1 and DDB2 | Binds specific DNA damage | XPE—mild skin sensitivity |
| XPA | Damage sensor | XPA—skin and neurologic problems: the most severe XP |
| XPC | Damage sensor | XPC—skin, tongue, and lip cancer |
| XPE | Damage sensor | XPE—neurologically normal |
| PRR | ||
| POLH | Polymerase | XPV—Mild to severe skin sensitivity; neurologically normal |
| Other syndromes | ||
| NER | ||
| Cockaynes | ||
| CSB | ATPase | Cutaneous, ocular, neurologic, and somatic abnormalities; short stature, progressive |
| deafness, mental retardation, neurologic degeneration, early death; sometimes presents together with XPB | ||
| Juberg-Marsidi | ||
| ATRX | Putative helicase | Thalassemia/mental retardation |
| SB | ||
| Nijmegen | ||
| NBS1 | Nibrin, cell-cycle regulation | Microencephaly; mental retardation; immunodeficiency; growth retardation; radiation sensitivity; predisposition to malignancy |
| Ataxia-telangiectasia | ||
| ATM | Phosphorylation | Neurologic deficiencies, manifest by inability to coordinate muscle actions; skin and corneal telangiectases. Leukemia, lymphoma, and other malignancies (breast cancer?) |
| MRE11 (Ataxia-like) | Exonuclease | DNA damage sensitivity; genomic instability; telomere shortening; aberrant meiosis; severe combined immunodeficiency |
| PRKDC | Ser/Thr kinase | SCID |
| Bloom’s | ||
| BLM | DNA helicase | High rate of spontaneous lymphatic and other malignancy; high rate SCE |
| Fanconi anemia | ||
| FANCA-G | Protein control | Multiple congenital malformations; chromosome breaks; pancytopenia. |
| Telomere shortening | ||
| Werner | ||
| WRN | DNA helicase/exonuclease | Premature senility, short stature, exonuclease rapidly progressing cataracts, loss of connective tissue and muscle, premature arteriosclerosis, increase risk of malignancy |
| RecQ4 | DNA helicase | Osteosarcoma; premature aging |
Repair mechanisms: BER, base excision; DSB, double-strand break; HR, homologous recombination; MMR, mismatch; NER, nucleotide excision; PRR, postreplication; SB, strand break.
Diseases: HNPCC, hereditary nonpolyposis colon cancer; NHL, non-Hodgkin lymphoma.
Other abbreviations: SCE, sister chromatid exchange; SCID, severe combined immunodeficiency.
Direct DNA repair is affected by DNA alkyltransferases. These enzymes catalyze translocation of the alkyl moiety from an alkylated base (e.g., O6-methyldeoxyguanosine) to a cysteine residue at their active site in the absence of DNA strand scission. Thus, one molecule of the enzyme is capable of repairing one DNA alkyl lesion, in a suicide mechanism. The inactivation of this mechanism by promoter hypermethylation is associated with Kras G to A mutations in colon cancer.229
In DNA nucleotide excision repair, lesion recognition, preincision, incision, gap filling, and ligation are required, and the so-called excinuclease complex comprises 16 or more different proteins. Large distortions caused by bulky DNA adducts (e.g., BPDE-dG and 4ABP-dC) are recognized [xeroderma pigmentosum (XPA)] and removed by endonucleases (XPF, XPG, and FEN). A patch is then constructed (pol and pol e) and the free ends are ligated.
Base excision repair also removes a DNA segment containing an adduct; however, small adducts (e.g., 3-methyladenine) are generally the target so that there is overlap with direct repair. The adduct is removed by a glycosylase (hOgg1 and UDG), an apurinic endonuclease (APE1 or HAP1) degrades a few bases on the damaged strand, and a patch is synthesized (pol β) and ligated (DNA ligases: I, II, IIIa, IIIβ, and IV).
DNA mismatches occasionally occur, because excision repair processes incorporate unmodified or conventional, but noncomplementary, Watson–Crick bases opposite each other in the DNA helix. Transition mispairs (G-T or A-C) are repaired by the mismatch repair process more efficiently than transversion mispairs (G-G, A-A, G-A, C-C, C-T, and T-T). The mechanism for correcting mispairings is similar to that for nucleotide excision repair and resynthesis described earlier, but it generally involves the excision of large pieces of the DNA containing mispairings. Because the mismatch recognition protein is required to bind simultaneously to the mismatch and an unmethylated adenine in a GATC recognition sequence, it removes the whole intervening DNA sequence. The parental template strand is then used by the polymerase to fill the gap.
Double-strand DNA breaks can occur from exposure to ionizing radiation and oxidation. Consequences of double-strand DNA breaks are the inhibition of replication and transcription, and loss of heterozygosity. Double-strand DNA break repair occurs through HR, where the joining of the free ends is mediated by a DNA-protein kinase in a process that also protects the ends from nucleolytic attack. The free ends of the DNA then undergo ligation by DNA ligase IV. Genes known to code for DNA-repair enzymes that participate in this process include XRCC4, XRCC5, XRCC6, XRCC7, HRAD51B, HRAD52, RPA, and ATM.
Postreplication repair is a damage-tolerance mechanism and it occurs in response to DNA replication on a damaged template. The DNA polymerase stops at the replication fork when DNA damage is detected on the parental strand. Alternately, the polymerase proceeds past the lesion, leaving a gap in the newly synthesized strand. The gap is filled in one of two ways: either by recombination of the homologous parent strand with the daughter strand in a process that is mediated by a helical nucleoprotein (RAD51); or when a single nucleotide gap remains, mammalian DNA polymerases insert an adenine residue. Consequently, this mechanism may lead to recombinational events as well as base-mispairing.
Persistent nonrepaired DNA damage blocks the replication machinery. Cells have evolved translesion synthesis (TLS) DNA polymerases to bypass these blocks.230 Most of these TLS polymerases belong to the recently discovered Y family, have much lower stringency than replicative polymerases, and thus are error prone. An increased mutation frequency is an evolutionary trade-off for cellular survival.
Mutator phenotype
Cancer cells contain substantial numbers of genetic abnormalities when compared to normal cells. These abnormalities range from gross changes such as nondiploid number of chromosomes, that is, aneuploidy, and translocations or rearrangements of chromosomes, to much smaller changes in the DNA sequence including deletions, insertions, and single nucleotide substitutions. Therefore, carcinogenesis involves errors in (1) chromosomal segregation, (2) repair of DNA damage induced by either endogenous free radicals or environmental carcinogens, and (3) DNA replication. Loeb originally formulated the concept of the mutator phenotype in 197429, 60 to account for the high numbers of mutations in cancer cells when compared to the rarity of mutations in normal cells. Recent advances in the molecular analysis of carcinogenesis in human cells and animal models have refined the mutator phenotype61, 231 concept that is also linked to the clonal selection theory proposed by Nowell (Figure 4).232 Generally, the mutator phenotype hypothesis proposes that mutation rates in normal cells are insufficient to account for the large number of mutations found in cancer cells. Consequently, human tumors exhibit an elevated mutation rate that increases the likelihood of a tumor acquiring advantageous mutations. The hypothesis predicts that tumors are composed of cells harboring hundreds of thousands of mutations, as opposed to a small number of specific driver mutations, and that malignant cells within a tumor therefore constitute a highly heterogeneous population.231
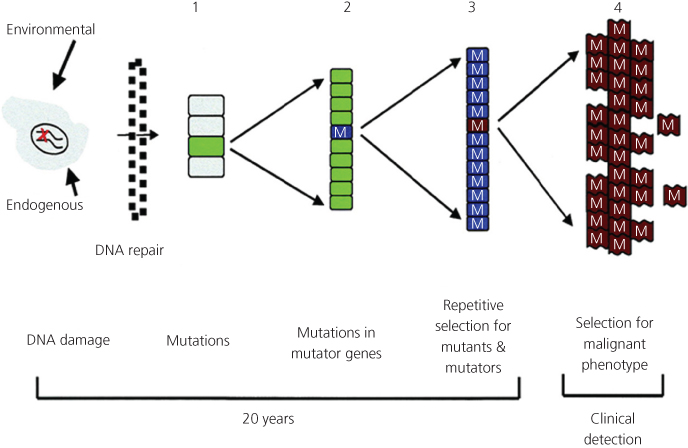
Figure 4 Mutation accumulation during tumor progression. (1) Random mutations result when DNA damage exceeds the cell’s capacity for error-free DNA repair. (2) These random mutations can result in clonal expansion and mutations in mutator genes (M). (3) Repetitive rounds of selection for mutants yield coselection mutants in mutator genes. (4) From this population of mutant cancer cells, there is selection for cells that escape the host’s regulatory mechanisms for the control of cell replication, invasion, and metastasis.
Racial, gender, and socioeconomic disparities in chemical carcinogenesis
Genetic polymorphisms may also partially explain the increased risk of cancer-associated chemical carcinogens amongst different genders, race, and ethnicity. One of the biggest controversies surrounding this issue is the risk of lung cancer in women smokers. The initial studies finding a higher risk of lung cancer in smoking women versus smoking men233 have been explained biologically in a number of ways. Several sources of evidence, though inconsistent, suggest that estrogens may play a role in lung cancer. Variation in risk factors and tumor characteristics between men and women has been reported in studies showing that women are more likely to have adenocarcinomas of the lung, a higher risk in never smokers, higher levels of PAH–DNA adducts, higher levels of expression of the gene encoding CYP P450 (CYP) 1A1, more frequent G : C to T : A transversions in p53, and more frequent epidermal growth factor receptor (EGFR) mutations than men.187 Inherited genetic polymorphisms affecting activating and detoxifying enzymes could also explain a different susceptibility between the sexes to tobacco carcinogens.234 In addition, several lifestyle and behavioral factors related to smoking habits or environmental and occupational exposures could account for some sex differences.235 However, in recent years, differences in the susceptibility to lung cancer among smoking men and women have been called into question.236, 237
A consistent theme is that nonsmoking women are at increased risk for lung cancer compared to nonsmoking men237, 238; and nonsmoking women are at increased risk for lung cancer due to environmental tobacco smoke (ETS) compared to nonsmoking women not exposed to ETS.239–244 This risk may be associated with polymorphisms. For example, in female populations, it appears that a common genetic polymorphism divides the population of never smokers into two groups of approximately equal size, one (homozygous carriers of the GSTM1 null allele) that has a statistically significant greater risk of lung cancer from ETS than the other (heterozygous or homozygous carriers of the wild-type GSTM1 allele).240 In a similar context, genetic variants at 13q31.3 alter the expression of the glypican-5 (GPC5) gene, and down-regulation of GPC5 might contribute to the development of lung cancer in never smokers.245, 246 Other genes that have been found in never smokers and associated with lung cancer risk are LEM domain-containing protein 3 (LEMD3), transmembrane Bax inhibitor containing motif (TMBIM), ataxin 7-like 2 (ATXN7L2), Src homology 2 domain containing E (SHE), inter-α-trypsin inhibitor heavy chain H2 (ITIH2), Nudix (nucleoside diphosphate linked moiety X)-type motif 5 (NUDT5), EGFR, and echinoderm microtubule-associated protein like 4–anaplastic lymphoma kinase (EML4–ALK) fusions.243, 247, 248 Similarly, α(1)ATD carriers are at a 70–100% increased risk of lung cancer.249 Finally, as is the case for p53, the mutational profile of k-ras seems to differ greatly according to smoking status: G to T transversions are more common in ever smokers, whereas G to A transitions are seen more frequently in never smokers. K-ras and EGFR mutations are generally mutually exclusive, underlining their ordered participation in an intracellular signaling pathway, and suggesting different oncogenic mechanisms in lung cancer as a function of smoking status: tobacco carcinogens seem to be directly responsible for k-ras mutations, whereas the cause of mutations in EGFR, found particularly in never smokers, is currently not clear.
For race, it appears that compared to Caucasians, African-Americans are diagnosed earlier, have more aggressive, and consequently, have lower survival rates from breast cancers.250, 251 Racial disparities also exist in colorectal cancer.252 Although these differences can be explained by socioeconomic differences, screening behaviors, and other behavioral differences, biological influences can affect risk. For example, polymorphisms in nucleotide excision repair genes may modify the relationship between breast cancer and smoking in African-American women.253 As well, the chemical carcinogen detoxifying gene, GSTM1, has a polymorphism that plays a role in the development of lung cancer and modifies the risk for smoking-related lung cancer in African-Americans.254 Interesting recent findings highlighting gene–environment interactions indicated household income is associated with the p53 mutational frequency in patients who develop breast cancer (especially ER-negative). Furthermore, high-income patients may acquire fewer p53 mutations than other patients, suggesting that lifetime exposures associated with socio-economic status may impact breast cancer biology.255, 256
Chronic inflammation and cancer
More than a century ago, the German pathologist, Virchow proposed that inflammation was associated with cancer.8 Infection and inflammation significantly contribute to more than 25% of cancer cases worldwide (Table 4).204 Free radicals, endogenous chemicals, are released during the inflammatory response. These ROS and reactive nitrogen species are generated as a physiological protective response to pathogenic microorganisms and toxic agents. During chronic inflammation (e.g., chronic viral hepatitis) and oxyradical overload conditions (e.g., hemochromatosis and inflammatory bowel diseases), these free radicals can induce genetic and epigenetic changes including somatic mutations in cancer-related genes and post-translational modifications in proteins involved in DNA repair, apoptosis, and arachidonic acid cascade (Figure 5).8 Epigenetic transcriptional silencing of cancer-related genes including p16, Runt-related transcription factor 3 (RUNX3), and MutL homolog 1 (MLH1), by DNA methylation of their promoter regions has been associated with chronic inflammation in ulcerative colitis and Barretts esophagus.257, 258 Inflammation-mediated genetic and epigenetic alterations can also drive cancer development in the neighboring epithelium upon stromal abrogation of transforming growth factor-β signaling.259 Finally, polymorphisms in genes involved in the production of [e.g., COX2, NOS’s (NOS1, NOS2; but mostly NOS3)] or protection from (e.g., GpX, catalase, and MnSOD) free radicals also exist; can modify the effects of drugs; and increase or decrease cancer risk associated with exogenous chemical carcinogens.260–268
Table 4 Chronic inflammation and infection can increase cancer risk
| Disease | Tumor site | Risk |
| Inherited | ||
| Hemochromatosis | Liver | 219 |
| Crohn’s disease | Colon | 3 |
| Ulcerative colitis | Colon | 6 |
| Acquired | ||
| Viral | ||
| Hepatitis B | Liver | 88 |
| Hepatitis C | Liver | 30 |
| Bacterial | ||
| Helicobacter pylori | Gastric | 11 |
| PID | Ovary | 3 |
| Parasitic | ||
| Schistosoma hematobium | Urinary bladder | 2–14 |
| Schistosoma japonicum | Colon | 2–6 |
| Liver fluke | Liver | 14 |
| Chemical/physical/metabolic | ||
| Acid reflux | Esophagus | 50–100 |
| Asbestos | Lung pleural | >10 |
| Obesity | Multiple sites | 1.3–6.5 |
“18% of human cancers, that is, 1.6 million per year, are related to infection.”—B. Stewart and P. Kleihues, World Cancer, Report, IARC Press, Lyon 2003, p. 57.
Rheumatoid arthritis is an example of a chronic inflammatory disease without a marked increased cancer risk, for example, joint sarcoma.
Oncogenic human papilloma viruses are examples of cancer-prone chronic infections without inflammation.
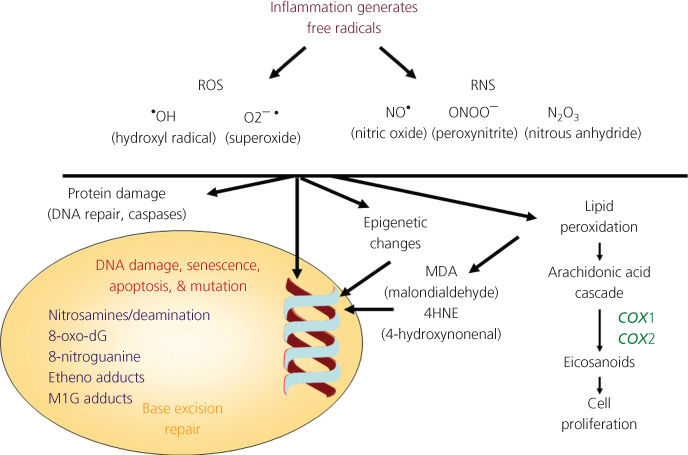
Figure 5 Several reactive oxygen species (ROS) and reactive nitrogen species (RNS) are generated during chronic inflammation. The reactive species can induce DNA damage, including point mutations in cancer-related genes, and modifications in essential cellular proteins that are involved in DNA repair, apoptosis, and cell cycle, either directly or indirectly through the activation of lipid peroxidation and generation of reactive aldehydes, for example, malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE).
Oncogenes and tumor suppressor genes
Chronic exposures to carcinogens, accumulation of mutations, development of the mutator phenotype, and clonal selection during several decades result in cancer. Although the phenotypic traits of individual cancers are highly variable, commonly acquired capabilities include limitless replicative potential, self-sufficiency in growth signals, insensitivity to antigrowth signals, evading apoptosis, tissue invasion, sustained angiogenesis, and metastasis.64 These phenotypic traits reflect a complex molecular circuitry of biochemical pathways and protein machines within cancer cells.269
The genes encoding the proteins within the cancer-associated molecular circuitry are of many functional classes and, historically, have been conceptually divided into oncogenes and tumor suppressor genes269, 270 (Box 4). Detailed descriptions of oncogenes and tumor suppressor genes are found in Chapters 4–7. The ras oncogene and the TP53 tumor suppressor gene will be used as examples of molecular targets of chemical carcinogens.
Box 4 Mutational hotspots in cancer genes
Oncogenes and suppressor genes are targets of chemical carcinogens. Many genes, including K-ras and p53, have specific sequences targeted by chemical carcinogens resulting in mutational “hotspots.” These mutations cause a change in expression and activity of the RNA and protein they code for.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


