José G. Montoya, John C. Boothroyd, Joseph A. Kovacs
Toxoplasma gondii
Although Toxoplasma gondii infects a large proportion of the world’s human population, it is an uncommon cause of disease. Certain individuals, however, are at high risk for severe or life-threatening disease because of this parasite. These include congenitally infected fetuses and newborns and immunologically impaired individuals. Congenital toxoplasmosis is the result of maternal infection acquired during gestation, an infection that is most often clinically inapparent. In immunodeficient patients, toxoplasmosis most often occurs in persons with defects in T-cell–mediated immunity, such as those receiving corticosteroids, anti–tumor necrosis factor (TNF) therapies, certain monoclonal antibodies, or cytotoxic drugs, and in those with hematologic malignancies, organ transplants, or acquired immunodeficiency syndrome (AIDS). In the vast majority of otherwise immunocompetent individuals, primary or chronic (latent) infection with T. gondii is asymptomatic; after the acute infection, a small percentage suffer chorioretinitis; lymphadenitis; or, even more rarely, hepatitis, myocarditis, and polymyositis.1
T. gondii was first observed in the North African rodent (Ctenodactylus gundi) by Nicolle and Manceaux in 19082 and was recognized as a cause of human disease in an 11-month-old congenitally infected child by Janku in 1923.3 It was reported as a cause of encephalitis by Wolf, Cowen, and Paige, who in 1939 observed it in a newborn who presented with seizures, intracranial calcifications, hydrocephalus, and chorioretinitis.4 Although relatively few cases of severe toxoplasmosis in adults were reported during the ensuing years, the remarkable report in 1968 by Vietzke and his colleagues,5 from the National Cancer Institute of the National Institutes of Health, highlighted T. gondii as a cause of life-threatening infection in patients with malignancy, predominantly in those with hematologic malignancies. Brain involvement with focal areas of encephalitis was the primary finding at autopsy in these patients. Since that time, several hundred cases in non-AIDS immunodeficient patients have been recorded in the literature.6 In 1983, the first report of toxoplasmosis in AIDS patients appeared.7 Toxoplasmic encephalitis (TE) subsequently was recognized as the major cause of space-occupying lesions in the brains of these patients, almost all of whom had serologic evidence of prior exposure to the parasite.7 Despite the significant advances that have been achieved in the recent past, major challenges remain in the areas of prevention and management of the acute infection in pregnancy, the fetus, and the newborn,8 and in the understanding and treatment of toxoplasmic chorioretinitis9 and infection in immunocompromised individuals.1,6,10
Etiology
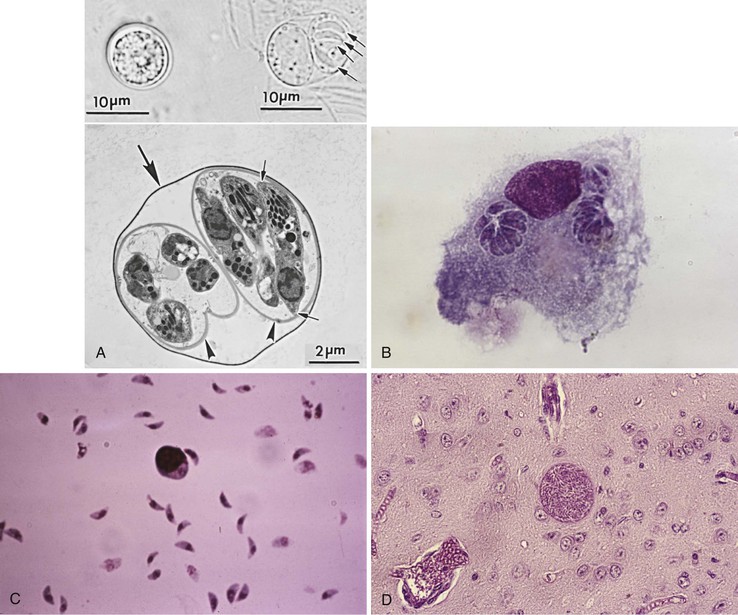
T. gondii is a coccidian parasite of felids with humans and other warm-blooded animals serving as intermediate hosts. It belongs to the subphylum Apicomplexa, class Sporozoa, and exists in nature in many forms: macrogametes and microgametes, the oocyst (which releases sporozoites), the tissue cyst (which contains and may release bradyzoites), and the tachyzoite (Fig. 280-1).11
Population genetic analysis has demonstrated that, at least within Europe and North America, most organisms isolated from both domesticated animals and humans can be grouped into one of three clonal genotypes—types I to III—that may identify clinically relevant biologic differences.12 Clear differences have been observed in the frequency of parasite genotypes when T. gondii isolates from animals were compared with those of humans. Type III strains are common in animals but observed significantly less often in cases of human toxoplasmosis; most cases in humans in Europe and North America are caused by type II strains. Type II strains are significantly more often associated with reactivation of chronic infections and accounted for 65% of strains isolated from AIDS patients.13 Both type I and type II strains have been associated with human congenital toxoplasmosis.13–15 To date, types II and III have not been detected in immunocompetent individuals with severe ocular disease.16 Atypical and recombinant strains have been identified with increasing frequency in regions other than the United States and Europe and from animals other than humans and domestic animals; some of these strains appear to be associated with more severe disease, suggesting greater virulence, even in immunocompetent individuals.17,18 The most exhaustive studies have now identified six major population clades,19 although detailed sequence analysis indicates there is a varied amount of genetic exchange within and between these clades.20 Hence, although the “three-dominant-strain” paradigm is holding for humans in Europe and North America, the situation appears much more complex in other regions and in other animal hosts.
Organism Stages
Oocyst
Cats eventually shed oocysts after they ingest any of the three forms of the parasite, at which time an enteroepithelial cycle begins. This sexual form of reproduction begins when the parasites penetrate the epithelial cells of the small intestine and initiate development of asexual and sexual (gametogony) forms of the parasite. Oocyst wall formation begins around the fertilized gamete, and when still immature, oocysts are discharged into the intestinal lumen by rupture of intestinal epithelial cells.11 Unsporulated oocysts are subspherical to spherical and measure 10 × 12 µm in diameter (see Fig. 280-1A). Oocysts are formed in the small intestine only in felids and are excreted in the feces for periods varying from 7 to 20 days. More than 100 million oocysts may be shed in the feces in a single day.11 Sporulation, required for oocysts to become infectious, occurs outside the cat within 1 to 5 days, depending on temperature and the availability of oxygen. Sporulated oocysts contain two sporocysts (see Fig. 280-1A), each of which contains four sporozoites. Maturation is more rapid at warm temperatures (2 to 3 days at 24° C compared with 14 to 21 days at 11° C).11 Oocysts may remain viable for as long as 18 months in moist soil; this results in an environmental reservoir from which incidental hosts may be infected. Recent clues to some of the features that make oocysts so robust have come from proteomic and transcriptomic analyses.21,22 Among other findings, these studies have revealed an abundance of small tyrosine-rich proteins in the oocyst wall. Cross-linking of the tyrosines in these proteins could confer a natural “sun-screen” for the sporozoites within because they are strong absorbers of ultraviolet light.
Tachyzoite
The tachyzoite form (see Fig. 280-1B) is oval to crescentic and measures 2 to 3 µm wide and 5 to 7 µm long; it requires an intracellular habitat to multiply despite having all of the usual eukaryotic machinery necessary for reproduction. Tachyzoites are seen in both primary and reactivated infection; their presence is the hallmark of active infection. They reside and multiply within vacuoles in their host’s cells, can infect virtually all phagocytic and nonphagocytic cell types,11 and multiply approximately every 6 to 8 hours to form rosettes.23 Continuous multiplication leads to cell disruption and release of organisms that go on to invade nearby cells or are transported to other areas of the body by blood and lymph.24 Tachyzoites appear to actively and rapidly migrate across epithelial cells and may traffic to distant sites while extracellular.25 Recent evidence suggests they might also use the infected host cell as a “Trojan horse” to traffic and gain access to tissues that might not otherwise be easily accessed.26
At the anterior end of the tachyzoite, there is a cone-shaped structure termed the conoid. It is protruded during the parasite’s entry into host cells. Rhoptries, numbering 4 to 12, are club-shaped organelles that terminate within the conoid. The rhoptries, together with surrounding small, rod-shaped organelles (micronemes), have important secretory functions for parasitic invasion. Dense granules are organelles distributed throughout the cytoplasm. Their contents are released into a vacuole, termed the parasitophorous vacuole, that is formed around the parasite during entry into the cell and also into the external environment as excreted-secreted antigens.11
The rhoptries and micronemes produce a collection of proteins that are crucial for the invasion process.27 These appear to mediate the attachment to the host cell, including the moving junction, a ringlike point of contact between the parasite and host cell surface that migrates down the length of the parasite during invasion. The rhoptries also inject proteins into the host cell that are critical in manipulating the host cell, presumably to the advantage of the parasite.28 These rhoptry proteins are very different (polymorphic) between different strains of T. gondii and appear responsible for many of the differences in virulence seen for types I, II and III in mice, as described earlier.
Tachyzoites cannot survive desiccation, freezing and thawing, or extended exposure to gastric digestive juices.24 They are propagated in the laboratory in the peritoneum of mice and in cultured cells. Tachyzoites can be visualized in sections stained with hematoxylin and eosin but are better visualized with Wright-Giemsa and immunoperoxidase stains.29
A fourth organelle that is restricted to Toxoplasma and its Apicomplexa cousins is the apicoplast.30 This is akin to chloroplasts of plants, with a similar evolutionary origin involving endosymbiotic algae, but photosynthetic functions have been completely lost. It has its own DNA, RNA, and protein translation, the latter of which is prokaryotic in nature, making for a very attractive target for drug therapies. Indeed, one of the currently used drugs for treatment of human infection, clindamycin, targets the ribosomes of the apicoplast.31 A major role of this organelle is fatty acid biosynthesis.30 Recent work with Plasmodium, which also has this organelle, has demonstrated that the apicoplast is crucial to the synthesis of isoprenoids,32 making further work on attacking this Achilles heel an attractive prospect.
Tissue Cyst
Once the tachyzoite has invaded the target cell, it can undergo stage conversion into the bradyzoite form.11 Tachyzoites and bradyzoites are structurally and phenotypically different. Tachyzoites multiply rapidly and synchronously, forming rosettes and lysing the cell, whereas the more slowly replicating bradyzoites form tissue cysts. Molecules are expressed in a stage-specific manner and are responsible for certain of the phenotypic differences between tachyzoites and bradyzoites. Interferon-γ (IFN-γ), nitric oxide (NO), heat shock proteins, and pH and temperature manipulations can trigger conversion of tachyzoites to bradyzoites in vitro and perhaps in vivo as well.11
Tissue cysts grow and remain within the host cell cytoplasm as the intracystic form wherein the bradyzoites continue to divide. Tissue cysts vary in size from younger ones that contain only a few bradyzoites to older tissue cysts that may contain several thousand bradyzoites and may reach more than 100 µm in size (see Fig. 280-1D). They appear spherical in the brain and conform to the shape of muscle fibers in heart and skeletal muscles. The central nervous system (CNS); eye; and skeletal, smooth, and heart muscles appear to be the most common sites of latent infection.33 Because of their persistence in tissues, demonstration of tissue cysts in histologic sections does not necessarily mean that the infection was recently acquired or that it is clinically relevant. Tissue cysts stain well with periodic acid–Schiff, Wright-Giemsa, Gomori methenamine silver, and immunoperoxidase stains. Tissue cysts in meat are rendered nonviable by γ-irradiation (0.4 kGy),34 heating meat throughout to 67° C, or freezing to −20° C for 24 hours and then thawing,35,36 but not by gentle heating in a microwave.37
Although the tachyzoite form appears to be indiscriminate in the type of host cell parasitized, it has been suggested that, in brain tissue, there is a predilection for tissue cyst formation to occur predominantly within neurons.38,39 However, it has been shown that tissue cysts can form within astrocytes cultured in vitro.40 In an electron microscopic study of the pathologic changes in brains of infected mice, tissue cysts were observed to remain intracellular throughout the period of study (22 months).38 There is compelling evidence to suggest that bradyzoites can exit from intact tissue cysts and invade contiguous cells, where they convert to the tachyzoite form.41 This is the likely explanation for the appearance of “daughter” cysts or clumps of cysts in the brain. Recent evidence suggests that neurons are capable of destroying the invaded parasite and/or that rhoptry proteins are injected into cells they do not infect.42 These results have implications for how the parasite commandeers host functions.
Transmission and Epidemiology
T. gondii infection is a worldwide zoonosis. The organism infects herbivorous, omnivorous, and carnivorous animals, including birds.43 Infection in humans most commonly occurs through the ingestion of raw or undercooked meat that contains tissue cysts, through the ingestion of water or food contaminated with oocysts, or congenitally through transplacental transmission from a mother who acquired her infection during gestation (Fig. 280-2). Less common are transmission by transplantation of an infected organ or transfusion of contaminated blood cells. Transmission has also occurred by accidental sticks with contaminated needles44 or through exposing open lesions or mucosal surfaces to the parasite.45 Because the sexual cycle of the parasite takes place in the small bowel of members of the cat family, cats play a significant role as powerful amplifiers of the infection in nature (see “Oocyst”).11 Epidemiologic surveys have revealed that in most areas of the world, the presence of cats is of primary importance for the transmission of the parasite. Excretion of oocysts has been reported to occur in approximately 1% of cats in diverse areas of the world.45 Wild felines, and especially bobcats in the United States, may be among the most important sources of oocysts.46
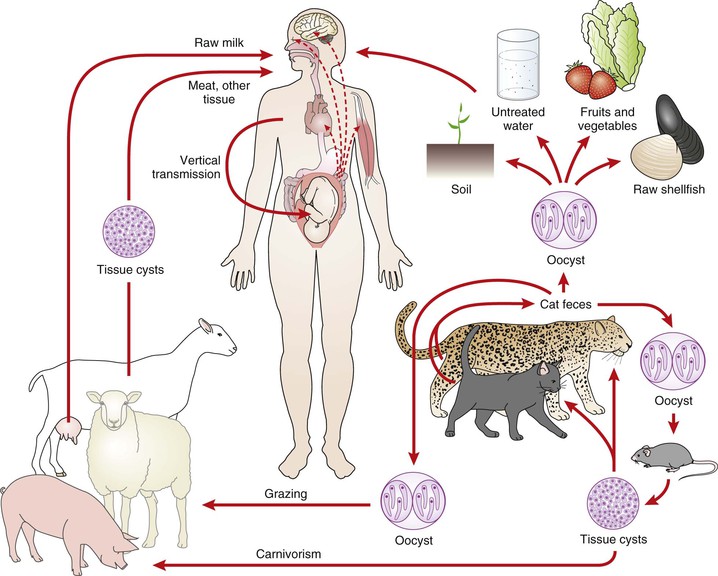
Although ingestion of raw or undercooked meat that contains viable T. gondii tissue cysts will result in infection, the relative frequency with which this occurs in relation to the frequency of infection caused by ingestion of oocysts is unclear. For instance, in countries such as France, where eating undercooked meat is common and the prevalence of the infection is high, meat may be an important cause of the infection. (It was in Paris that the meat-to-human hypothesis of spread of T. gondii was proved.47) In contrast are countries such as those in Central and South America, where the prevalence of the infection in humans is high but the ingestion of undercooked meat is uncommon. In these areas, oocysts may be the more important source of human infection.48 Until a newly described method for distinguishing bradyzoite- versus oocyst-initiated infection is validated in these regions,49 their respective contributions to human infection will largely remain a matter of speculation.
Ingestion of tissue cysts in infected meat (primarily pork and lamb) is a major source of the infection in humans in the United States.50,51 T. gondii infection is common in many animals used for food, especially sheep and pigs, with a lower prevalence in cattle, horses, and water buffaloes. Infection resulting in transmission to humans has also been documented in wild game animals.52 Organisms may survive in tissue cysts in these animals for years and can be found in nearly all edible portions of an animal.53 A seminal study on the prevalence of T. gondii in samples of meat used for human consumption (obtained from grocery stores) was performed in the United States in the 1960s.54 The parasite was isolated from 32% of pork chops and 4% of lamb chops; there were no isolations from beef, and indeed, cattle appear to be generally not an important intermediate host for this parasite.55 A recent polymerase chain reaction (PCR)-based study in England found 33% (19/57) of pork and 67% (6/9) of lamb samples positive for T. gondii DNA.56
Serologic surveys conducted in the past 20 years in the United States indicate that the prevalence of T. gondii in pigs is declining, with an overall prevalence of 2.6% in a recent National Animal Health Monitoring Survey.57 This reduction in T. gondii prevalence has been attributed to changing management practices and consolidation of pig production into large-scale operations. However, there are still many isolated small swine farms, including those that raise organic pigs, and the prevalence of T. gondii in these animals can be greater than 90%.58 Of note, meat for human consumption is not routinely inspected for T. gondii infection in the United States or elsewhere in the world.59 Seroprevalence of T. gondii infection in a study of lambs in the mid-Atlantic region was recently reported to be 27%.60 Although T. gondii infection of sheep is widely prevalent, the public health importance of this is unclear because, in the United States, meat from adult sheep is not usually used for human consumption.45 Reports of suspect transmission by unpasteurized goat’s milk have appeared.61,62 In addition to differences in how meat is cooked, the tendency for beef to harbor few, if any, cysts compared with lamb and pork may partly explain the differences in seroprevalence in the United States versus Europe; beef accounts for a much greater fraction of meat consumed in the United States compared with Europe, where lamb and pork are more popular.
T. gondii infection is prevalent in game animals, especially black bears (80% infected) and white-tailed deer, as well as in raccoons (60% infected).53 Infection in raccoons and bears is a good indicator of the prevalence of T. gondii in the environment because these animals are scavengers. Thus, wild animal meat can serve as a source of the infection for hunters and their families, especially when care is not taken while eviscerating and handling the game or when meat and other organs from these animals is served undercooked or uncooked.53
Although T. gondii tissue cysts may be found in edible tissues of chickens,63 poultry products are probably not important in the transmission of T. gondii to humans because they are usually frozen for storage and thoroughly cooked to avoid diseases that could be caused by contamination by other organisms.45 The parasite has been isolated from chicken eggs.64
The ingestion of vegetables and other food products contaminated with oocysts probably accounts for infection in seropositive vegetarians. Although isolation of tachyzoites from secretions of people with the acute infection has been claimed, human-to-human transmission of infection by this route has not been established. Outbreaks within families and other groups are common,65–67 but there is no evidence of natural human-to-human transmission other than from mother to fetus.
Several epidemiologic studies have identified water as a potential source for T. gondii infection both in humans and animals.65,68–71 In vitro studies have demonstrated that oocysts can sporulate in seawater within 1 to 3 days, can survive in seawater for up to 6 months, and can survive in water treated with sodium hypochlorite or ozone, but not ultraviolet radiation.72–74 Population mapping studies of acutely infected individuals as well as case-control studies linked drinking unfiltered water (presumably contaminated with oocysts) to an outbreak of toxoplasmosis in a municipality in the Western Canadian province of British Columbia65 and to high endemic rates of toxoplasmosis in Rio de Janeiro State, Brazil.69 In another Brazilian outbreak, T. gondii organisms were detected in water by a variety of methods from an implicated reservoir.75 Coastal freshwater runoff was observed to be a risk factor for T. gondii infection among southern sea otters along the California coast.76
In humans, the incidence of T. gondii antibodies increases with increasing age; the incidence does not vary significantly between sexes. The incidence tends to be less in cold regions, in hot and arid areas, and at high elevations. Slaughterhouse workers may have an increased risk for infection. The prevalence of antibody titers to T. gondii varies considerably among different geographic areas and also among individuals within a given population. These differences depend on a variety of factors, including culinary habits and cleanliness of surroundings. A decrease in antibody prevalence over the past few decades has been observed in many countries. In the United States, the seroprevalence in U.S. military recruits decreased by one third between 1965 and 1989; the crude seropositivity rate among recruits from 49 states was 9.5% in 1989 compared with 14.4% in 1965.77 As another example, in the 1970s, 24% of women in the childbearing age group in Palo Alto, California were seropositive, whereas the rate in 2008 was 10%. Seroprevalence rates in the United States among such women range from 3% to greater than 35%, whereas rates greater than 50% are present in women of childbearing age in much of Western Europe, Africa, and South and Central America.78 The recent (1999 to 2004) overall age-adjusted seroprevalence of T. gondii infection in the United States, based on a study of 15,960 persons aged 6 to 49, was reported to be 10.8%, with a seroprevalence among women aged 15 to 44 years of 11%.79 This study found an approximately 25% to 40% decline in seroprevalence compared with a similar survey a decade earlier. Although the prevalence of the infection appears to be declining in certain areas of the world, such as Europe and the United States, this has not been the case or it has been documented to have increased in other geographic locales.78
T. gondii may survive in citrated blood at 4° C for as long as 50 days, and infection has been transmitted through transfusion of whole blood or white blood cells. Leukocyte transfusions may pose a special risk.80 The transmission of infection by organ transplantation has been documented and may result from the transplantation of an organ (e.g., heart) from a seropositive donor to a seronegative recipient.81 In bone marrow transplant recipients, toxoplasmosis almost always is a result of recrudescence of a latent infection rather than from the transplant.82,83
The incidence of TE among HIV-infected individuals directly correlates with the prevalence of T. gondii antibodies among the general HIV-infected population, the degree of immunosuppression (best measured by the CD4 cell count),84 the use of effective prophylactic treatment regimens against development of TE, and the immunologic response to antiretroviral therapy (ART).85 AIDS-associated TE and toxoplasmosis involving other organs are almost always due to reactivation of a chronic (latent) infection that results from the progressive immune dysfunction that develops in these patients.86 It is estimated that 20% to 47% of AIDS patients who are infected with T. gondii but are not taking anti-Toxoplasma prophylaxis or antiretroviral drugs will ultimately develop TE.84,86 This makes TE a major concern in areas where the use of antiretrovirals is still a treatment relatively few HIV-positive individuals receive.
In recent years a substantial decline in the incidence of TE87 and toxoplasmosis-associated deaths88 has been seen in HIV-infected patients who adhere to effective anti-Toxoplasma prophylactic regimens and to ART.
In the United States, T. gondii seropositivity among HIV-infected patients varies from 10% to 45%86 and directly correlates with the seropositivity in the general non–HIV-infected population. In contrast, the seroprevalence is approximately 50% to 78% in certain areas of Western Europe and Africa.89,90 In a study in France, 1215 (72.2%) of 1683 HIV-infected patients had serologic evidence of exposure to T. gondii.91 During the study period (1988 to 1995), the overall incidence of toxoplasmosis in this population was estimated to be 1.53 per 100 patient-years, with an increase from 0.68 per 100 patient-years in 1988 to 2.1 per 100 patient-years in 1992, and a subsequent decline to 0.19 per 100 patient-years in 1995 that was likely related to the widespread use of anti-Toxoplasma prophylaxis. Toxoplasmosis is rare in the HIV-infected pediatric population: 0.06 cases per 100 patient-years were reported among more than 3000 patients participating in clinical trials in the pre-ART era but during a time when Pneumocystis carinii pneumonia (PCP) prophylaxis was recommended.92
Of interest is the low reported incidence of TE in Africa, despite T. gondii seroprevalence rates of 32% to 78%. Lack of autopsy data and a lack of neuroimaging studies likely contribute to the low reported incidence. It has also been suggested that because of poor access to medical care, many HIV-infected patients in Africa succumb to infection with organisms such as Mycobacterium tuberculosis before they develop the opportunistic infections associated with the advanced stage of HIV infection, including toxoplasmosis. However, in one autopsy series, from the Ivory Coast, of 175 patients with AIDS-defining abnormalities, the prevalence of TE was 21%.93
T. gondii infection may be acquired after the acquisition of HIV infection. Seroconversion rates between 2% and 5.5% have been reported in patients followed for periods up to 28 months.94
Even before the emergence of AIDS, TE had been recognized as a cause of incapacitating disease and death among HIV-negative immunosuppressed patients,6,95 especially in those whose underlying disease or therapy caused a deficiency in cell-mediated immunity. Patients with hematologic malignancies, especially those with Hodgkin’s disease, are at a particularly higher risk to develop recrudescence of the infection. Among organ transplantation patients, those with heart, lung, kidney, and bone marrow transplants develop toxoplasmosis at a higher rate.
Pathogenesis and Immunity
T. gondii multiplies intracellularly at the site of invasion (the gastrointestinal tract is the major route for and the initial site of infection in nature); bradyzoites released from tissue cysts or sporozoites released from oocysts penetrate, differentiate to tachyzoites, and then rapidly multiply within intestinal epithelial cells. Organisms may spread first to the mesenteric lymph nodes and then to distant organs by invasion of lymphatics and blood. T. gondii tachyzoites infect virtually all cell types, and cell invasion occurs as an active process. Survival of tachyzoites is due to the formation of a parasitophorous vacuole that lacks host proteins necessary for fusion with lysosomes,96 and consequently acidification does not occur. Active invasion of macrophages by tachyzoites does not trigger oxidative killing mechanisms. With the appearance of humoral and cellular immunity, only those parasites protected by an intracellular habitat or within tissue cysts survive. An effective immune response significantly reduces the number of tachyzoites in all tissues, and after the initial acute stages, tachyzoites are rarely demonstrable histologically in tissues of infected immunocompetent humans. Tachyzoites are killed by reactive oxygen intermediates,97 acidification,98 osmotic fluctuations, reactive nitrogen intermediates,99 intracellular tryptophan depletion,100 and specific antibody combined with complement.101 In rodents, two classes of immune-stimulated guanosine triphosphate (GTP)ases play a crucial role in destruction of tachyzoites within the parasitophorous vacuole: the p47 immunity-related GTPases (IRGs)102 and the larger GTP-binding proteins (GBPs).103
Tissue cyst formation takes place in multiple organs and tissues during the first week of infection. Despite the ability to isolate T. gondii from normal brains of chronically infected humans, the tissue cyst form is rarely observed in histologic preparations; it has been isolated from both brain and skeletal muscle in 10% of 52 T. gondii–seropositive patients who, at autopsy, had no clinical or pathologic evidence of the infection.33 The tissue cyst form is responsible for residual (chronic or latent) infection and persists primarily in the brain, skeletal and heart muscle, and the eye.33,104
In immunocompetent individuals, the initial infection and the resultant seeding of different organs leads to a chronic or latent infection with little, if any, clinical significance. This chronic stage of the infection corresponds to the asymptomatic persistence of the tissue cyst form in multiple tissues. It is believed that periodically, bradyzoites are released from tissue cysts or that cysts “rupture”; cyst disruption in this setting appears to be a clinically silent process effectively contained by the immune system and in the CNS likely results in small inflammatory nodules, with a limited degree of neuronal cell death and architectural damage.41 However, several investigators have suggested that chronic infection may not be completely asymptomatic and may result in important behavioral changes and neuropsychiatric disorders,105 although definitive studies to support such associations have not yet been reported.
Toxoplasmosis in severely immunodeficient individuals may be caused by primary infection or be the result of recrudescence of a latent infection. It is widely held that reactivation is the result of disruption of the tissue cyst form, followed by differentiation to and uncontrolled proliferation of tachyzoites and tissue destruction. In individuals with deficient cell-mediated immunity, rapid, uncontrolled proliferation of T. gondii results in progressively enlarging necrotic lesions. It has been postulated that damage to any organ in these patients, including the brain, eye, heart, lung, skeletal muscle, gastrointestinal tract, and pancreas, can result directly from tissue cyst disruption in the parenchyma of the organ itself or from tissue cyst disruption elsewhere in the body, followed by subsequent spread to that organ.106 Hematogenous spread is supported by the observation of the development of simultaneous lesions in the brain and the presence of parasitemia in 14% to 38% of AIDS patients with TE.107,108
Infection with T. gondii induces both humoral and cell-mediated immune responses. A well-orchestrated and effective systemic immune response, combining both innate and adaptive mechanisms, is responsible for the early disappearance of T. gondii from peripheral blood during the acute infection and limits the parasite burden in other organs. Immunity in the immunocompetent host is lifelong. Exogenous reinfection, which has been demonstrated in laboratory animals, likely also occurs in humans but does not appear to result in clinically apparent disease, although, in one case report, a chronically infected pregnant woman was infected with a highly virulent strain that resulted in infection of the fetus.109
Because T. gondii is a natural parasite of rodents, inbred mice have been used extensively as an animal model for studies of both immunity and immunopathology in this protozoan infection and have yielded a remarkably detailed picture of its host interaction.110–112 When tachyzoites invade, they inject the contents of their rhoptries into the host cell cytosol. This delivers not only some of the machinery needed for invasion (contained within the rhoptry necks and known as RON proteins) but also a collection of “effectors” that intercept or co-opt host immune pathways, presumably to the parasite’s advantage. These effectors include the rhoptry protein, ROP16, which functions as a mimic of host Janus kinases (JAKs), phosphorylating critical tyrosines on host STATs (signal transducers and activators of transcription). Depending on the particular flavor of ROP16 that a given strain carries, the host immune response can be driven in differing directions, either more or less inflammatory, and this can have a profound effect on the host’s response and ultimate outcome of the infection. In the case of another pair of injected ROPs, ROP5 and ROP18, the target is murine IRGs, a key part of a mouse cell’s defense machinery.113 Normally, IRGs attack the membrane of the vacuole in which a pathogen resides, disrupting it and leading to death of the organisms within. ROP5 and ROP18, however, collaborate to phosphorylate and thereby inactivate IRGs although, again, the effectiveness of this depends on the specific alleles of ROP5 and ROP18 that a given strain of Toxoplasma carries. Lastly, dense granules can also introduce polymorphic effectors into the host cell; one such effector, GRA15, has been shown to be crucial to the activation of one of the most central transcription factors in mammalian immune response, nuclear factor kappa B.114
It is important to recognize that these findings do not necessarily represent the mechanisms underlying the immune response to T. gondii in humans. For example, although the effect of ROP16 on STATs may well have a parallel in human cells, IRGs are not part of the human immune response, and so ROP5 and ROP18, if they are impacting the outcome of human infection, must be doing so by acting on other targets, such as activating transcription factor-6β.115 Similarly, Toll-like receptors TLR11 and TLR12 play a major role in the induction of interleukin-12 (IL-12) and host resistance in the mouse, but neither receptor exists in humans, indicating that innate recognition of the parasite in humans must involve distinct mechanisms. This might involve another branch of the innate immune system, NLRs (nucleotide oligomerization domain [NOD]-like receptors), that detect molecular signatures or patterns specific to various pathogens. Recent work has suggested that susceptibility to Toxoplasma infection in humans is associated with a polymorphism in a human NLR known as NALP1 (NACHT-LRR-PYD domains–containing protein 1).116 The overall question of how Toxoplasma is sensed by the innate arm of the human immune system and how different strains of the parasite do this with differing degrees of success is just beginning to be explored.
T cells, macrophages, and type 1 cytokines (IFN-γ, IL-12) are crucial for control of T. gondii infection. Adoptive transfer and depletion experiments in murine models not only proved that T cells are essential for control of T. gondii infection but also demonstrated an interplay between CD4+ and CD8+ T cells in both the induction of resistance and the maintenance of latency. Expansion of both natural killer (NK) and γδ T cells early in infection provides innate resistance while the adaptive response mediated through αβ CD4 and CD8 T cells develops. These different subsets of T cells and NK cells are likely to protect the host by secreting cytokines, such as IFN-γ, IL-2, and TNF-α, and apparently not by lysing T. gondii–infected cells.117–120 Dendritic cells and inflammatory monocytes also play an important role in control of acute infection and the early production of IL-12.120,121 NK-cell–derived IFN-γ appears critical to the differentiation of IL-12–producing dendritic cells.120 Early studies suggested that neutrophils were an important component of the early response, but more recent studies suggest that they are not and may, in fact, contribute to the pathology.121
The co-stimulatory molecules CD28 and CD40 ligand are pivotal for the regulation of IL-12 and IFN-γ production in response to the parasite.122 T. gondii infection of antigen-presenting cells, such as dendritic cells and macrophages, causes upregulation of the counter-receptors for CD28 and CD40L, CD80/CD86, and CD40.122 Binding of CD80/CD86 to CD28 enhances production of IFN-γ by CD4+ T cells. In addition, binding of CD40L to CD40 triggers IL-12 secretion, which in turn enhances production of IFN-γ. The relevance of CD40L in the immune response to T. gondii is supported by reports of TE and disseminated toxoplasmosis in children with congenital defects in CD40L signaling (hyper-IgM syndrome).123 Moreover, recent studies have demonstrated that expression of CD40L is defective on CD4+ T cells from HIV-infected patients.124 This deficiency may play a role in defective IL-12/IFN-γ production associated with HIV infection.
Cytokines play a critical role in defense against the infection and are important in the pathogenesis of toxoplasmosis and TE.125 IL-12 enhances survival of T-cell–deficient mice during T. gondii infection, by stimulating the production of IFN-γ by NK cells,126 and is thought to also regulate the expression of the latter cytokine by T cells in immunocompetent mice.127 IFN-γ has been shown to play a significant role in the prevention or development of TE in mice.128 The administration to chronically infected mice of a monoclonal antibody against IFN-γ resulted in a dramatic worsening in the degree of encephalitis.129 In mice with active TE, treatment with IFN-γ significantly reduced the inflammatory response and numbers of tachyzoites.130
Differences in IL-12 levels elicited during infection by different strains of the parasite may be responsible for some of the strain-specific differences in virulence in mice.131 These differences appear to be related to the activation (phosphorylation) of the transcription factor STAT3, which in turn is dependent on the particular allele of ROP16 injected by a given strain, as detailed earlier.132
TNF-α is another cytokine pivotal for control of T. gondii infection. TNF-α is required for triggering of IFN-γ–mediated activation of macrophages for T. gondii killing activity133 and for nitric oxide (NO, an inhibitor of T. gondii replication) production by macrophages.134 The administration of TNF-α neutralizing antibody to infected mice caused the death of the mice and an increase in the number of T. gondii tissue cysts in the brains of survivors.135
IL-10 has been shown to deactivate macrophages and result in reduced in vitro killing of T. gondii. IL-4 and IL-6, which are usually considered downregulatory cytokines, have been shown to be important in resistance against TE in the murine model.136,137 IL-7 has also been shown to have a protective role against T. gondii in mice.138 During the early stages of the infection, IL-12, IL-1, and TNF act in concert with IL-15 to stimulate NK cells to produce IFN-γ.139
IL-17140 and IL-23141,142 have also been implicated in the generation of a potent immune response but are not thought be essential for host resistance.
Several hypotheses have been proposed to explain the role of IFN-γ in host resistance to T. gondii. Involvement of reactive nitrogen intermediates (including NO) is suggested by the observation that l-NG-monomethyl-l-arginine acetate (l-NMMA), a competitive analogue of l-arginine, simultaneously inhibits NO synthesis and intracellular tachyzoite killing by cytokine-activated peritoneal macrophages and microglial cells.99,143,144 In addition, mice in which NO synthesis is impaired as a result of genetic disruptions of the IFN-γ or IFN-1 genes succumb to the acute infection.145,146 Similar enhanced susceptibility was observed in mice treated with the reactive nitrogen intermediate inhibitor aminoguanidine147 and in nitric oxide synthase–deficient mice.148 The protective role of NO appears to be tissue-specific rather than systemic.148 Because control of the acute infection in vivo was unaffected by nitric oxide synthase deficiency, the major role of reactive nitrogen intermediates appears to be to maintain control of established infections in this mouse model.148
The IFN-γ–inducible p47 GTPases IRGM3 (IGTP) and IRGM1 (LRG47) have been shown to be required for host control of T. gondii infection in the mouse,149 and recent studies have linked IRGM3 with the autophagic destruction of Toxoplasma-containing vacuoles in IFN-γ–activated macrophages.150,151
Immunoglobulin G (IgG), IgM, IgA, and IgE antibodies are produced in response to the infection. Extracellular tachyzoites are lysed by specific antibody when combined with complement. In mice, humoral immunity results in limited protection against less virulent strains of T. gondii but not against virulent strains.152
Both astrocytes and microglia likely play important roles in the immune response against T. gondii within the CNS. In the early stages of TE in both humans and mice, there is a remarkable and widespread astrocytosis restricted to areas in which the parasite is detected.125 Whereas T. gondii can invade, survive, and multiply within astrocytes, they are killed by activated microglia.153
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


